HMMER通常与已构建好的HMM数据集一起使用,例如Pfam以及Interpro收纳的一些数据库。同时HMMER也可以像BLAST一样使用查询序列,对序列数据库(非HMM数据库)进行检索。例如,您可以使用phmmer将一条蛋白质序列检索序列数据库(类似于BLASTP了),也可以用jackhmmer进行迭代检索。
HMMER依靠其潜在概率模型的强度,旨在尽可能敏感地检测远程同源序列。
HMMER可以用来在数据库中搜索蛋白或DNA序列的同源物,并且做序列比对。HMMER可以仅仅用来在数据库中查询序列,但因为可以在做多重序列家族的比对而变得更加强大。HMMER给替换,插入。删除突变设置了一个位置特异性评分系统的查询文件。HMMER框架是一个叫做“隐形马尔可夫模型”的概率性模型。
跟BLAST,FASTA和一些序列比对和基于老的评分方法的数据库搜素相比,HMMER的目的是更准确和更能删掉同源物,因为他强大的概率模型。在过去,这个优势是需要很强高的电脑计算能力的配置,在蛋白质和DNA搜索的过程中,他的速度比BLAST慢100倍。但现在的HMMER3.1在蛋白搜素上的速度跟BLAST差不多,同时在DNA搜索中也仅比BLAST慢5-10倍。
HMMER可以作为命令行工具进行本地下载和安装,现在也可以通过EBI的服务器在线访问。
本地下载地址:http://hmmer.org/
在线版本:http://www.ebi.ac.uk/Tools/hmmer/
常用Pfam在线数据库:http://pfam.xfam.org/
HMMER包含下面几个主要的程序:
phmmer: 与Blastp类似,使用一个蛋白质序列搜索蛋白质序列库;
jackhmmer: 与psiBlast类似,蛋白质序列迭代搜索蛋白质序列库;
hmmbuild: 用多重比对序列构建HMM模型;
hmmsearch: 使用HMM模型搜索序列库;
hmmscan: 使用序列搜索HMM库;
hmmalign: 使用HMM为线索,构建多重比对序列;
hmmconvert: 转换HMM格式
hmmemit: 从HMM模型中,得到一个模式序列;
hmmfetch: 通过名字或者接受号从HMM库中取回一个HMM模型;
hmmpress:格式化HMM数据库,以便于hmmscan搜索使用;
hmmstat: 显示HMM数据库的统计信息;
常用的两大功能
(一)使用HMM数据集搜索全基因组蛋白(核酸)序列数据库
1、hmmbuild, 训练给定多序列比对结果,构建HMM
数据集。举个例子像在基因家族分析中,用所有已知的某基因家族成员做多序列比对,然后利用下面命令构建HMM数据集,最后使用HMM数据集扫描你要鉴定的物种所有基因序列数据库即可获得获得该物种候选的该基因家族成员。
示例命令:
hmmbuild [-options]
输入文件 多 序 列 比 对 后 的 序 列 格 式 如:CLUSTALW, SELEX, GCG MSF。
输出文件一般命名为.hmm 后缀, 该结果为HMM 数据库。
2)hmmsearch, 寻找相似序列
hmmsearch [options]
(二)使用蛋白质(核酸)序列搜索已构建HMM数据库
该方法为常用的功能注释方法。
构建HMM数据库。使用多序列比对文件,同上述命令即可完成构建。同时可以从Pfam、SMART等网站下载现成额HMM。举个例子,假如我有一批蛋白质序列,想做Pfam注释,看看有什么结构域,那么我可以去Pfam下载下述文件:
ftp://ftp.ebi.ac.uk/pub/databases/Pfam/releases/Pfam31.0/Pfam-A.hmm.gz
使用hmmscan搜索HMM数据库,命令如下:
hmmscan -E 0.00001 --domE 0.00001 --cpu 2 --noali --acc --notextw --domtblout pfam.tab Pfam-A.hmm test.pep.fa
输出结果介绍
主要介绍两种格式
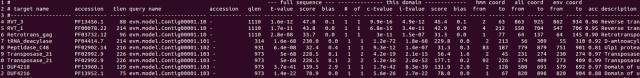
--domtblout
--tblout
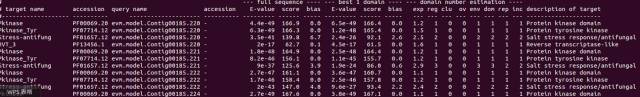
输出结果中分为两类一类是针对序列的(full sequence) ,另一类是针对domain的(主要基于一条序列存在多个domain)。这两种格式涉及到的每一列信息解释如下(英文原文大家看的可能更明白!)
(1) target name: The name of the target sequence or profile.
(2) accession: The accession of the target sequence or profile, or ’-’ if none.
(3) query name: The name of the query sequence or profile.
(4) accession: The accession of the query sequence or profile, or ’-’ if none.
(5) hmmfrom: The position in the hmm at which the hit starts.
(6) hmm to: The position in the hmm at which the hit ends.
(7) alifrom: The position in the target sequence at which the hit starts.
(8) ali to: The position in the target sequence at which the hit ends.
(9) envfrom: The position in the target sequence at which the surrounding envelope starts. 结构域的起始位置。
(10) env to: The position in the target sequence at which the surrounding envelope ends. 结构域的终止位置。
(11) sq len: The length of the target sequence..
(12) strand: The strand on which the hit was found (“-” when alifrom¿ali to).
(13) E-value: The expectation value (statistical significance) of the target, as above.










