
►
药品改革,与我们每个人息息相关,图片来自china.org.cn
编者按:
5月21日,在2017中国国际药物信息大会暨第九届DIA中国年会上,国家食品药品监督管理局(
CFDA
)国际合作司司长袁林透露,中国加入ICH的时机基本成熟,已提交了有条件地加入ICH的申请。ICH 全称为International Council for Harmonization,旨在协调不同国家间药品质量、安全性和有效性的技术规范,推动药品注册要求的一致性和科学性。这则重磅消息联合5月11-12日CFDA发布的四个“征求意见稿”,几乎是将中国医药产业置于全球格局之中参与竞争,提高中国老百姓用药的可及性和质量。与此同时,这也必将对中国整个医药行业及相关方产生深刻的影响。
撰文 | 谢雨礼
责编 | 叶水送
●
●
●
近年来,食品安全、药品价格高、看病难等问题一直为老百姓所诟病。中国医药行业能不能提高产品质量和医疗服务水平,满足人们日益增长的健康需求?
2017年3月17日,国家食品药品监督管理局
(CFDA)
发布《关于调整进口药品注册管理有关事项的决定(
征求意见稿
)》,降低国外新药进入中国的政策门槛,实现中国新药研发和上市与全球同步。
正当业内还在讨论“狼来了”该怎么办
(担忧跨国公司在新政的鼓励下,加快进入中国的步伐,并且占据有限的临床试验资源,不利于国内本土药企创新)
。5月11日-12日,CFDA两天连发四个以“鼓励”开头的文件
(意见征求稿)
。这是自2015年CFDA启动改革以来,鼓励创新力度最大的一次,触及当前掣肘药物创新的多个方面,包括审评审批速度慢、临床试验水平低和资源不足等问题。
事实上,从一致性评价、临床数据打假、全球同步到这次的4个“鼓励文件”,信号已非常明确,那就是监管与国际接轨,为加入ICH做准备。最近,CFDA官员在第九届DIA中国年会上表示,中国将有条件地加入ICH。
这意味着,CFDA改革围绕医药产品的供给侧全面展开。以毕井泉局长为首的CFDA领导层态度坚决,改革的力度和效率前所未有,两年来发布了一系列里程碑式的政策和措施(图1)。这次改革的方向主要包括4个方面:1)加快审评审批速度;2)提高产品质量;3)鼓励创新;4)降低价格。
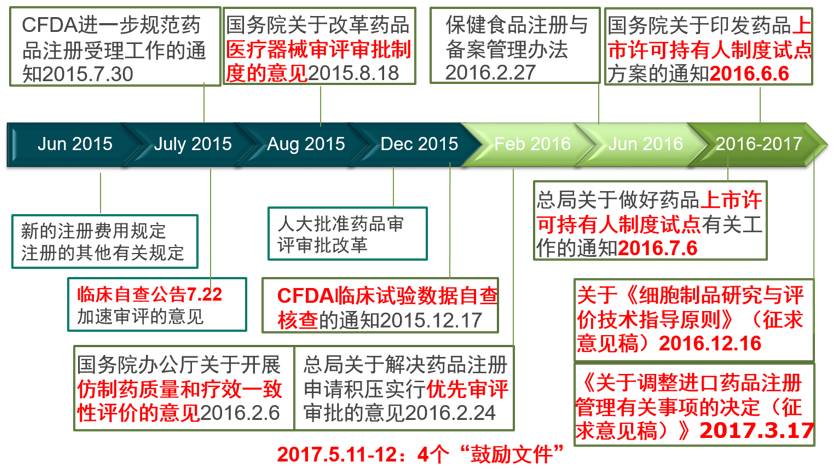
►
图1:2015-2017年CFDA改革的里程碑事件
过去中国药品审评审批速度非常慢、积压严重。从药学研究开始算,一个仿制药的开发需要7-10年时间,其中临床批件和生产批件的审评时间加起来超过4年。对于新药而言,中国的上市时间平均比欧美发达国家要慢4-5年。中国实行医保制度,新药上市后进医保又需要另一个4-5年。这样,中国普通老百姓享受可支付新药速度平均比美国晚8年(
2009医保目录分析报告
)。
中国药品审评速度慢有几个客观原因。首先是国内医药行业集中度低,有几千家药厂,低水平重复申请严重,药审中心每年要收到几千件申请,其中绝大多数为重复申请的仿制药;第二,药审中心的人手和经费不足,过去中心的工作人员只有120人左右,而美国FDA的同等机构工作人员多达5000人以上;第三,中国过去缺乏优先审评的路径,也没有政策遏制重复申请,造成新药和临床急需药,与仿制药同时排队审批。
针对这些情况,CFDA近年采取了一系列改革措施,加快审评审批速度。为解决积压严重的问题,实行集中审评,已清理完数万件积压申请。人手方面,通过公开招聘,扩充一线审评队伍。据了解,2017年底,药审中心工作人员将增至600人。另一方面通过借调地方工作人员和外部专家参加集中审评,购买第三方服务,缓解短期人手以及经验不足的困难。
2015年,CFDA出台政策重新定义化学仿制药和新药(
如图2
),改革后将新药分为新化学实体和改良新药,而仿制药分为仿国外和仿国内两种情况。新的定义和分类更加符合国际惯例。

►
图2:改革前后化药的定义和分类
基于新的定义CFDA设计了十大优先审评情况,鼓励创新、解决临床急需以及提高药品质量。规定具有明显临床价值的创新药,重大疾病治疗药物,如恶性肿瘤,临床短缺药物,如儿童药,欧美认可的高质量药物等,均获得优先审评的资格。同时还通过出台各种措施、简化和规范审评流程,比如上调仿制药申请费用、抑制重复申请等等。
国产药品质量不如进口药或原研药几乎已经成为老百姓的常识。因此,国人在国外旅游时,顺便购买药品也成了一道风景。从整体上来说,中国仿制药质量的确不如原研药,但我们也应注意到每个品种,情况有所差异。保证药品质量是监管机构的职责之一。过去,我国仿制药审批的标准低,只要符合国家标准即可,并不严格要求与原研药一致,而且对申报材料中数据的完整性和真实性监管不严。行业内的人都知道,过去生物等效试验的通过率几乎达到100%。
为提高药品质量,CFDA从监管的角度出发,采取了三大改革措施:1)仿制药一致性评价;2)临床数据和工艺自查和核查;3)审评和审批体系升级。
仿制药一致性评价是药品供给侧改革的重大举措。行业改革,通过国务院高规格发布具体意见和政策,非常罕见,体现了其重要性。仿制药一致性评价,美国和日本等制药强国都经历过这一步。仿制药达到与原研药一致,就能实现临床互换,有利于降低药品价格。按照政策要求,基药中2007年以前批准的口服品种必须在2018年10月1日之前通过一致性评价,否则吊销药证。
如果容许数据做假,任何严格的要求和高标准将会成为摆设。2015年7月22日,CFDA发起了临床数据自查和核查。对数据做假零容忍,涉及做假的企业、临床基地、CRO公司以及直接责任人,将受到严厉查处。“7.22临床核查”风暴发现大面积临床数据不合规或造假,造成80%以上的申请自撤或被撤,数十亿的研发投入作废,对业界影响巨大。如果说临床核查主要是针对在研品种的话,不出意外,CFDA下一步将会积极推进上市品种的生产工艺核查。可以预计,将有一批企业和上市品种受到影响,甚至被停产或吊销药证。
审评和审批体系升级包括提高药品的审批标准和上市药品再注册的标准,建立与国际接轨的审评流程和标准化程度。另外,还需要建立一支专业化的技术审评队伍,明确审评人员的权利和责任(
终身负责
)。为了达到这些改革的目标,除了自身努力,CFDA还尝试了依靠外部力量。2016年6月13日,CFDA 宣布与盖茨基金合作,引进国际审评专家,比如CFDA首席科学家何如意博士。据了解,今年还会大力引进科学家,提高审评队伍的专业能力和经验,保证审评的质量和速度。
中国95%以上的药品为仿制药。过去几十年,中国自己研发的1.1类新药,也就是新化学实体(
NCEs
)约30个,其中大部分缺乏真正的临床价值,也少有商业成功。最近,随着埃克替尼、西达本胺和阿帕替尼等新药上市,情况有所改善。令人担忧的是,中国在研创新药,以me-too为主,差异化不足,出现“替尼大战”和PD-1大战等“高水平重复”现象。无论如何,这轮改革,鼓励创新是重要的主题。
创新药,特别是全球新的NCE, 在审评上享受绿色通道,优先度排在第一位。另外,在10省市率先试点上市许可人制度(
MAH
),意味着科研人员和院所可以在没有GMP工厂的情况下持有药证,有利于创新和资源优化。当然,如何做到主体责任明晰,控制药品质量风险以及建立药品质量保险制度,仍在积极探索当中。
5月中旬,CFDA发布的4个“鼓励”文件,支持创新的力度前所未有:
在
52
号文中,将加快新药上市的政策具体化,包括有条件批准临床急需药品和罕见病药物、原辅料和包装材料备案和联合审评、与审评机构的沟通渠道、支持新药进入医保等。
在
53
号改革临床试验管理的文件中,着手解决临床试验资源短缺的矛盾,放开临床试验机构的认定,改为备案制,并支持研究者和临床试验机构开展临床试验。另外,提出完善伦理委员会机制和提高伦理审查效率。特别重要的一条是,规定审评机构自受理临床试验申请之日起
60
个工作日后,没有给出否定或质疑的审查意见即视为同意,申请人可按照递交的方案开展临床试验,虽然较
FDA
的
30
个工作日仍有差距,但相对于以前动辄
2
年的等待时间已是巨大的进步。文件还提出接受境外临床试验数据作为申报依据,会同此前公布的进口药品注册的改革,将进一步加速国外药品进入中国的速度。支持拓展性临床试验,则是对应于
FDA
的同情用药,显示了监管机构的弹性。规定严重危及生命且尚无有效治疗手段疾病的药物医疗器械,经临床试验初步观察可能获益,且符合伦理要求的,经知情同意后可用于其他未参加试验的患者。
54
号文涉及药品和器械的生命周期管理政策,包括上市许可人法律责任,不良反应报告制度,药品和器械再评价,打假及规范学术推广,加强监管和检查等。中国是注射剂大国,在价格和市场扭曲的驱动下,存在滥用现象。这里特别值得关注的是开展上市注射剂再评价。与
52
号文中严格注射剂审评审批的规定相呼应。目前仍不明确是否涉及中药注射剂的再评价。
55
号文对药品专利链接制度和试验数据保护制度等保护创新者权益的政策作出了规定,具体内容类似于
FDA
的相关政策,势必进一步激发创新者的热情。
纵观这些新政,很多涉及其他部门,比如支持新药进医保、改革临床试验管理等,不是CFDA独自能够落地执行。因此,如何协调是个难题。或许类似于一致性评价,上升到国务院层面解决,更加合理。
中国某些药品存在价格虚高的情况。主要是一些所谓单独招标和单独定价的品种,包括专利过期的原研药。一份研究报告显示,非专利抗肿瘤药物的月费用,中国在全球排第一,超过欧美发达国家,亦远超人口差不多的印度(
图3
)。
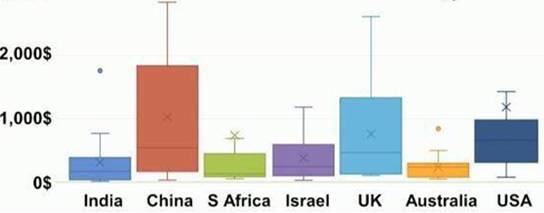
►
图3:15种主要的非专利抗肿瘤药物的月费用平均值(2016 ASCO研究报告)
药品价格虚高的原因很多。首当其冲的是我国“以药养医”的医疗体系,医院和医生的收入取决于药品销售,造成药品越贵越好卖的怪象。加之,药品销售不规范,存在多级中间商,回扣现象普遍。招标和定价政策执行变样,也是推广药价的因素。各地招标政策,五花八门。招标的本意是控制价格、鼓励创新、鼓励高质量。然而在利益面前,好的政策都流于形式,执行时变味,药品价格越招越高。政府部门决定商品价格,必然滋生腐败,成为推高药价的黑手。
中国和成熟市场的药品价值链差异很大。以美国为例,研发和生产药品的药企,占到价值链的70%,也就是100块钱的药品,企业要拿70元,而药批和药房只有30元。而中国,药企只能拿到20%左右,各级中间商和医院以及其他灰色地带占到80%。显然,这十分不利于药企的创新和发展。目前,医疗改革推行的“两票制”和医院零加成就是针对这一弊端。
有人可能会认为,降低价格应该是卫计委和招标办的责任,与CFDA改革没什么关系。这就错了,要知道,所有招标、定价、医保和进医院等政策,都是以药品的名义制定的。比如只要是新药就能获得招标和定价的优势。这就是为什么,中国一度出现为了新药而新药的现象,片剂改胶囊、葡萄糖中添果糖,市场上新药满天飞。这次改革,特别定义了改良新药,规定没有明显临床优势的不批准。注射剂的价格比口服贵,销售空间大,因此中国市场对注射剂上瘾,莫名其妙出现了一批疗效不明确的注射剂,畅销市场。一些成分不明的中药注射剂,一度还带来大量安全事故。
令人高兴的是,这次4个鼓励文件中,特别提到了严格注射剂审评审批和再评价。另外,加快审批和仿制药一致性评价,也将对药品价格产生巨大影响。如果国产药品通过一致性评价,原研药就失去了单独招标和单独定价的法理基础。下一步一定是招标改革,原研药将会与国产药品PK价格。最后,鼓励创新的政策,会加快新靶点me-too药物上市的速度,这对原研专利药的价格也会造成重大冲击。当然,我们应该保持清醒的头脑,不能为了降低价格而降低标准。因此,我们的仿制药和me-too药物能否突破原研的技术壁垒和专利,保持至少相当的疗效和质量,变得至关重要。
这轮改革可以说是CFDA历史上最深刻的一次变革。改革的目的是供给侧结构转型,及时地为老百姓提供创新产品,高质量和经济可及的医药产品。改革将会推动中国医药市场走向成熟,各项政策与国际接轨,整个行业融入全球,互通有无。
►
表1:改革前后各项政策对比
|
政策法规
|
美国或其他成熟市场
|
中国现状
|
改革
|
|
申请人
|
可以是自然人/可委托加工
|
申报和生产企业捆绑
|
MAH试点;鼓励创新
|
|
技术要求
|
ICH指导原则;非常完善
|
逐渐完善, follow ICH
|
日趋严格,对研发质量要求高
|
|
审批流程
|
一报一批;简单明晰
|
繁琐,不科学(BE需审批)
|
BE备案;临床大批件
|
|
审批周期
|
1-2年
|
5-7年
|
加快审评速度;鼓励创新;改革临床试验管理等
|
|
专利
|
专利挑战(180天市场独占权)
|
取消专利到期两年前申报的限制
|
专利链接制度。数据保护
|
|
产品质量
|
仿制类似原研,可自由替换
|
与原研有差距
|
一致性评价;数据打假
|
|
产品价格
|
仿制药大幅下降
|
原研不受专利过期影响
|
改革市场准入和招标
|
|
销售模式
|
大型分销商;分销商毛利为主要激励机制
|
多层级分销商和自然人(挂靠);激励机制扭曲
|
改革销售模式,打击商业贿赂;回归产品和质量竞争
|
|
入市速度
|
快;大型分销商
|
慢;批准,招标和进医院
|
改革市场准入
|
|
原研替代
|
快;6个月内
|
慢;原研长期占优
|
取消原研优势;提高国产质量
|
|
行业竞争
|
集中度高
|
低端分散,竞争激烈
|
淘汰低水平;保证产品质量
|
对于传统药企来说,这轮改革最大的冲击是仿制药一致性评价。特别是基本药物中289个品种,已经划下生死线,2018年必须通过,涉及14000个文号和2000多家企业。一致性评价绝不是做做测试这么简单,绝大多数品种、药学不过关。所谓一致性评价,就是重新开发一个仿制药,难度非常大。面对挑战,大多数药企并没有准备好,抱怨甚多。大意是“昨天我还在街上卖早点,怎么一夜之间就要按五星级饭店来要求”。
另一大影响,就是临床核查造成大量研发投入作废。过去几年,许多药企非理性投入3类和6类药研发。“7.22临床核查”造成大量申请撤回,研发投入作废。同时,手上积攒大量原3.1类临床批件,进退两难,成为鸡肋。
这轮改革下,许多传统药企迷失了方向。其实,改革发出的讯号已给传统药企指明了发展方向,那就是三条路:1)高质量仿制药;2)海外市场;3)创新药。但由于仿制药竞争激烈,海外市场准入困难,创新药成功率低,一批低水平的中小企业不可避免地面临淘汰,特别是那些品种单一,没有自己营销渠道的企业。
然而,改革也给创新和研发能力强的企业带来了机遇。首先,一致性评价和质量升级将淘汰低水平重复,市场上同品种的厂家减少,可以保证一定利润。另外,一致性评价后,原研单独招标和定价失去合理性,必将释放一部分市场份额给通过一致性的本地企业。长期垄断市场的原研产品,大多有技术壁垒,比如缓控释、特殊注射剂等。这既是挑战,也是机遇。
中国研发能力和审批标准的提高,必将提高中国药品海外市场的竞争力。企业花费大量人力物力通过BE后, 也愿意尝试中美双报。一旦中国通过ANDA的产品多起来,形成了组合,议价能力就能提升,有利于打开美国等海外市场。华海和恒瑞等大企业已在国外布局多年,有望率先突破。
一致性评价政策出台后,国内一片哗然,认为这是有利于跨国公司的举措。然而,有远见的外企并不这样认为。显然,如前所述,一致性后,原研药降低价格,原研非专利药市场将逐步减少。贝恩咨询公司预测中国市场将走向成熟,逐步从专利药、原研非专利药和仿制药组成的三级结构,变成类似于欧美的二级结构(
图4
)。如果跨国公司不改变现有的策略,5-6年内原研非专利药将会变得无利可图。事实上,已有跨国公司意识到了这一点,采取措施应对改革,比如阿斯利康去年将其两个原研产品依姆多和波依定出售给国内药企,从而更加聚焦于专利药。有的跨国公司则通过产品授权和技术合作,与国内药企进行战略合作。
同时,进口药品注册改革和接受国外临床数据等举措,将会加快跨国公司的专利药进入中国市场的速度。阿斯利康非小细胞肺癌药物奥希替尼(
Tagrisso
)
以创纪录的速度在中国上市,就是一个很好的开端。
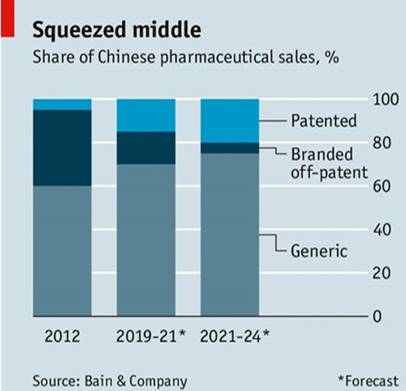
►
图4:贝恩咨询公司预测中国市场的变化
如上分析,改革鼓励和有利于创新,这一点无容置疑。然而,改革进口药品注册和这次的4个“鼓励”文件,将中国尚在起步阶段的创新研发一下推入全球的格局之中,可以说是前有狼后有虎。我们应该如何创新,做什么样的创新变得微妙起来。改革将会对生物医药创新公司的选题方向和商业模式产生重要影响。
中国大多数病人,长期以来,难以及时地享受到最新和最好的药品。除了新药上市速度慢,没有商业保险,患者支付能力差,也是重要原因。因此,me-too药物上市有利于降低新药的价格,提高药品的可及性,具有重要的意义。通过价格的差异化,也能取得市场的成功,贝达的埃克替尼就是一个很好的例子。即使今天,跨国公司的原研产品加快进入中国,只要支付能力的差异存在,me-too药物仍然有所作为。
然而,与过去不同的是,me-too药物的研发变得竞争非常激烈,风险也大大增加。一个热门的靶点,开发中国市场的药企众多。以抗肿瘤药物PARP抑制剂为例,既有阿斯利康的首创药物olaparaib,也有再鼎引进的TESARO开发的Niraparib,还有百济神州、其他传统药企和科研院所开发的数个产品,均已获得临床批件。
风险既包括上市后的市场风险,又包括研发风险。市场风险,不言而喻,因为对手众多。另外,“再鼎模式”在全球同步和数据互认等政策的鼓励下,将成为常态,必然会加速首创的第一梯队产品进入中国,让国内me-too竞争雪上加霜。研发风险来源于CFDA审评审批与国际接轨标准的提高。原研加速上市,后续开发较晚的me-too极有可能要求头对头比较。既然这样,具有临床优势或差异化的研发策略变得非常重要。
最近几年,中国基础研究发展很快。科研院所中已培育了一批原创的、具有新药开发潜力的苗子。在日益活跃的资本支持下,这些成果正在按国际惯例进行转化。中国原创已经起步,今后10年,必然迎来高速发展。中国与国际接轨,既为国外药品进中国打开了大门,也为中国创新药走向世界铺平了道路,从事原创研究的生物医药公司前景可期。当然,中国公司独立开发的可能性不大,更多是转让海外权益或者与海外公司合作的模式。过去两年,中国转让海外权益,已有不少案例(
表2
)
。
►
表2:中国2015-2016海外权益转让案例
|
Licensor
|
Licensee
|
date
|
assets
|
stage
|
upfront
|
total
|
|
信达
|
Lilly
|
2015.3
|
PD-1
|
Discovery
|
$56M
|
$1B
|
|
恒瑞
|
Incyte
|
2015.9
|
PD-1
|
Phase I
|
$25M
|
$795M
|
|
康方生物
|
Merck
|
2015.12
|
AK-107
|
Discovery
|
NA
|
$200M
|
|
正大天晴
|
J&J
|
2016.1
|
Anti-HBV
|
Discovery
|
NA
|
$253M
|
|
复旦大学
|
沪亚
|
2016.3
|
IDOi
|
Discovery
|
NA
|
$67M
|
就创新而言,有产业和销售渠道支持的传统药企,可以瞄准中国市场,开展me-too药物的研发。也可以从投资的角度,适当探索原始创新。生物医药公司,特别是风投支持的小公司,则要瞄准全球,具有足够的创新性,才可吸引到战略合作伙伴或更多的资本。
这轮改革对于CRO公司是重大利好,仅一致性评价就会带来50亿的相关业务,包括药学研究和生物等效试验等,临床试验管理临床CRO业务将迎来井喷式的发展。另外,上市人许可制度给CMO业务创造大量机会。鼓励创新,会让更多的药企借力CRO公司的平台,并发挥CRO作为国内外天然桥梁的作用,走国际化路线。当然,国际化和提升标准要求也给CRO公司提出更高的要求,因此小的CRO公司不一定能够适应。
巧合的是,有一个数据显示,中国生物医药风投比较活跃的2006年和2015年,刚好是CFDA最近两次改革的元年(
Dow Jones Venture Source
),这也说明,政策环境对于提升投资者的信心非常重要。中国现在是生物医药最好的时代,代表着一个巨大的风口。就需求来说,日渐富裕的中国人对健康的要求越来越高,就供给侧而言,无论是高质量的仿制药,还是新药,都与发达国家有差距,这其中蕴含着巨大的机会。
此次CFDA改革必将载于中国医药产业发展的史册,因为触动多方利益。从成立部级联席会议的举措可以看出,这次高层决心很大,不会像以前那样屈服地方保护主义。改革不但不会停,还会继续深化。也有悲观者认为,可能会雷声大雨点小。毕竟,现在发布的各项政策,都是框架性的顶层设计,落地执行还将面临无数具体问题,这也绝不是CFDA一个部门能够独立完成的任务,需要全民参与。改革不是发几个文件和意见就能一蹴而就的。比如一致性评价,也许企业在高压下会想方设法通过。问题是在日常生产中,能不能持之以恒,保证质量。
这次DIA会议上,日本同行透露,日本一致性评价运动中有10%的品种被吊销药证。但是在通过评价后5年的飞行检查中,只有0.43%不合格。要让企业保持产品质量,除了监督检查,更重要的是要通过市场回报机制,让企业主动去做。创新也是一样,没有市场回报的创新是不能持续的。
改革还要注意利益的平衡。比如,为了降低药价,改革以药养医的体制,但我们不能忘了当初实行这一制度的出发点,是为了解决医生的收入和医疗投入不足的矛盾。不解决医生和医院的诉求,改革断然不会成功。制药企业也是如此,没有合理的药品价格保证利润,就无法创新和保证质量。
改革仍在继续,大势不可违。受改革冲击的各方应采取措施,提高自己的核心竞争力。改革大潮下,不是发展就是被淘汰,没有别的选择。

参考文献
1)文中有些内容来自CFDA公开发布的政策和意见
2)有些观点和数据来自网络,仅供参考。





