催化氧化反应在化学工业中有着重要的应用,例如汽车尾气中污染物的三效催化与氢燃料电池中的CO优先氧化。优异的低温氧化催化剂需要氧活化-溢出和反应物活化的协同兼顾,这对活性中心的局部结构以及活性金属与催化剂载体之间的相互作用要求极高。负载型催化剂在汽车尾气净化中起着重要作用,但是传统的浸渍或沉积方法通常会导致活性金属完全分散在载体的顶部,弱的金属-载体相互作用限制了它们的最大性能。
针对这个极具挑战的合成难题,浙江大学化工学院
谢鹏飞团队
通过常规沉淀法和两步还原(聚集)-再分散法合成了CeO
2
稳定的Cu二聚体催化剂。通过建立各种Cu-CeO
2
组合的计算模型,利用相图进行从头算热力学分析,解析了铜二聚体的原子结构。综合电镜和光谱表征,结合DFT计算揭示了Cu在CeO
2
上的再分散过程伴随着Cu二聚体的形成,其中一个Cu原子嵌入CeO
2
晶格中,相邻的Cu原子被锚定在CeO
2
表面。得到的双中心Cu/CeO
2
-He催化剂由于其独特的几何结构和Cu
a
-Cu
b
双原子中心的协同效应,对各种催化氧化反应都表现出优异的催化性能。这项工作突出了气体预处理在双原子催化剂制备中的巨大潜力,可以广泛应用于高级氧化催化剂的活性中心调节。
该论文
第一作者为
研究生曹宁(浙江大学)、浦天宬博士(浙江大学)、戴升教授(华东理工大学)。
通讯作者为
浦天宬博士(浙江大学)和谢鹏飞研究员(浙江大学)。

在这项工作中,通过球差电镜发现Cu/CeO
2
催化剂在H
2
-He两步处理过程中Cu物种经理聚集-再分散的过程,最终得到的Cu/CeO
2
-He催化剂中Cu为原子级分散。通过原位光谱和从头算热力学分析进一步解析Cu/CeO
2
催化剂的原子结构发现Cu为独特的二聚体结构。双中心 Cu/CeO
2
-He 催化剂中Cu
a
被锚定在CeO
2
表面,呈现出Cu
1+
特征,Cu
b
嵌入CeO
2
的晶格之中呈现出Cu
2+
特征。
相对于Cu单原子和CuO团簇催化剂,Cu二聚体催化剂在CO优先氧化以及三效催化(CO氧化,C
3
H
6
氧化和NO氧化)中表现出更优的催化性能和100h的稳定性。DFT机理研究进一步表明,Cu/CeO
2
-He催化剂在CO氧化过程中,由于 Cu
a
-Cu
b
二聚体独特的协同效应,使 Cu/CeO
2
-He 催化剂在反应过程同时促进反应物的吸附和晶格氧的活化,克服了单原子催化剂和团簇催化剂无法兼顾实现反应物吸附和晶格氧活化的难题。
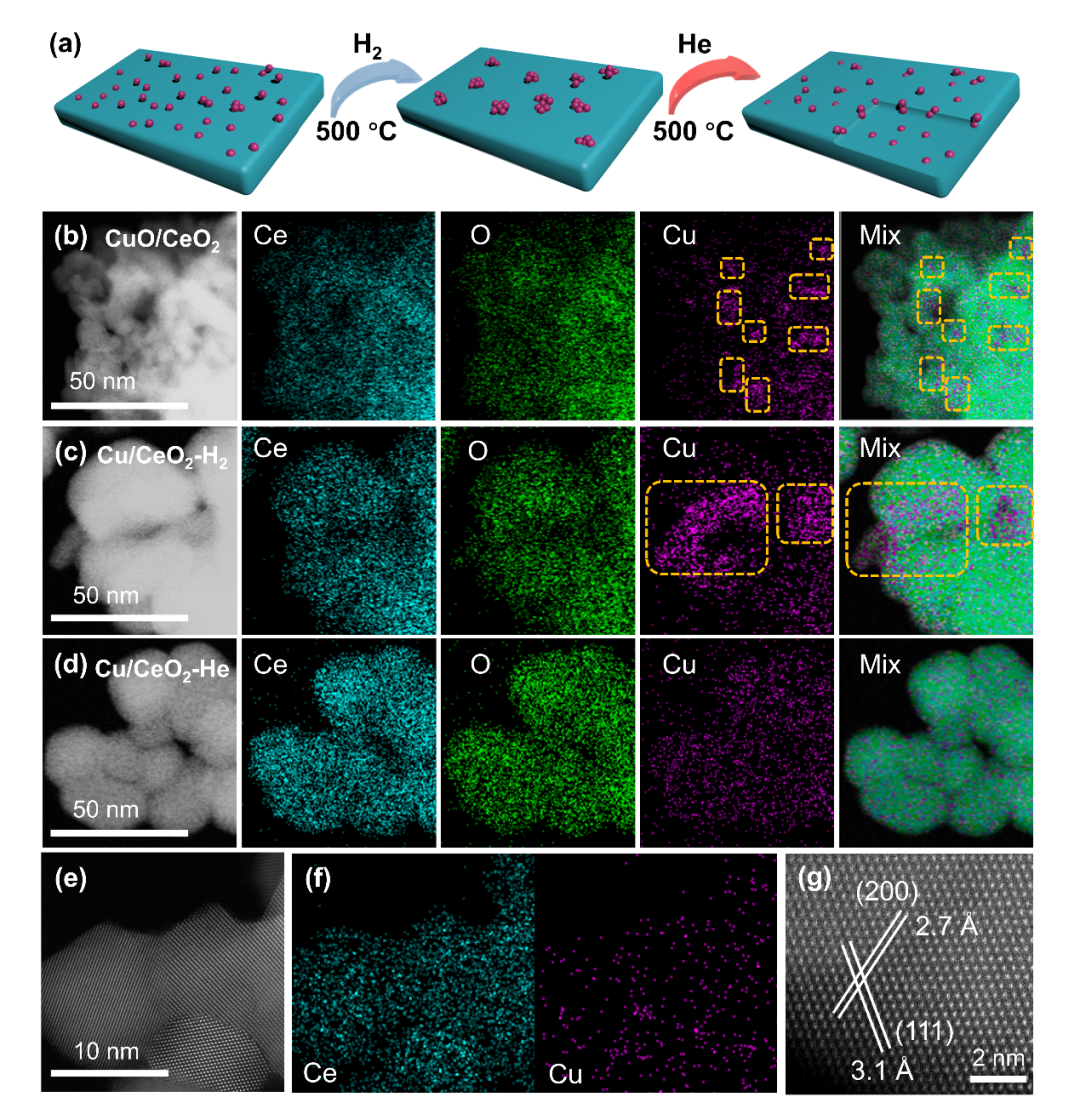
Figure 1. Synthesis of ceria supported Cu dimer catalysts.
(a) Schematic illustration of the catalyst synthesis process; STEM mapping results of CuO/CeO
2
(b), Cu/CeO
2
-H
2
(c), Cu/CeO
2
-He (d); (e-g) AC-HAADF-STEM image and STEM mapping of Cu/CeO
2
-He.
图1. CeO
2
负载Cu二聚体催化剂的合成及电镜结果。
通过两步法合成了高分散的Cu/CeO
2
催化剂。电镜结果表明在H
2
-He两步处理过程中Cu物种会经历聚集-再分散的过程,两步法得到的Cu/CeO
2
-He催化剂中Cu物种为原子级分散状态。

Figure 2. Characterization and modeling of the Cu/CeO
2
catalysts.
(a)
In situ
k
2
-weighted EXAFS spectra at the Cu K edge of Cu/CeO
2
after H
2
and He pretreatments and (b)
In situ
XANES spectra of Cu/CeO
2
after H
2
and He pretreatments, with Cu foil, Cu
2
O and CuO as reference samples. (c) Graphical illustration of various possible models with dispersed Cu species supported on CeO
2
; (d)
Ab initio
thermodynamic phase diagram estimating the stability of Cu species on CeO
2
; (e) Fitting of the EXAFS spectrum with on-dop-Cu
2
model. (f) Electrostatic potential calculation of Cu pair in on-dop-Cu
2
model.
图2. Cu/CeO
2
-He催化剂的原子结构解析。
XAS分析表明Cu/CeO
2
-He催化剂中Cu物种并未发生团聚形成Cu-Cu键,Cu的化学态在1+和2+之间角色塑造和模拟。从头算热力学计算表明在实验条件下on-dop-Cu
2
的二聚体结构最为稳定。DFT计算的结构与XAS的拟合结果一致,Cu/CeO
2
-He中的Cu物种不同与Cu单原子的Cu团簇,为二聚体结构。其中Cu
b
嵌入CeO
2
的晶格之中呈现出Cu
2+
特征,Cu
a
被锚定在CeO
2
表面,呈现出Cu
1+
特征。

Figure 3.Catalytic performance of Cu/CeO
2
-He catalyst compared to CuO/CeO
2
counterpart.
(a) Preferential CO oxidation in excess of H
2
; (b) CO oxidation; (c) Propene oxidation; (d) NO oxidation with O
2
; (e) Stability test of Cu/CeO
2
-He catalyst in aforementioned oxidation reactions in 100 h period.
图3. Cu/CeO
2
-He催化剂在CO优先氧化和三效催化(CO氧化,丙烯氧化和NO氧化)中与 CuO/CeO
2
催化剂的活性对比。
Cu/CeO
2
-He催化剂在CO优先氧化和三效催化中都表现出了更低的反应温度及更高的转化率,同时可以在100小时内保持活性稳定。

Figure 4.Catalytic cycle and free energy diagrams of CO oxidation over various model Cu/CeO
2
catalysts.
(a) Schematic illustration of catalytic cycle of CO oxidation over dimeric Cu supported on CeO
2
(Cu/CeO
2
-He); (b) Free energy diagrams of CO oxidation over Cu/CeO
2
-He, Cu single atom supported on CeO
2
(Cu
1
/CeO
2
) and CuO supported on CeO
2
(CuO/CeO
2
); (c) Comparison of the energy barriers of key transition states (TS-1 and TS-2) over Cu/CeO
2
-He, Cu
1
/CeO
2
and CuO/CeO
2
.
图4. 以CO氧化为探针反应研究了不同Cu/CeO2催化剂的氧化反应机理。CeO2负载的Cu双原子,Cu单原子以及CuO团簇均以MvK机理进行CO氧化,且第一个CO的氧化为整个反应的决速步骤。Cu双原子中Cu
b
促进了晶格氧的活化,而Cu
a
促进了CO的吸附,Cu
a
-Cu
b
之间的协同效应使得Cu双原子催化剂相比与Cu单原子和CuO团簇催化剂具有低的动力学反应能垒。
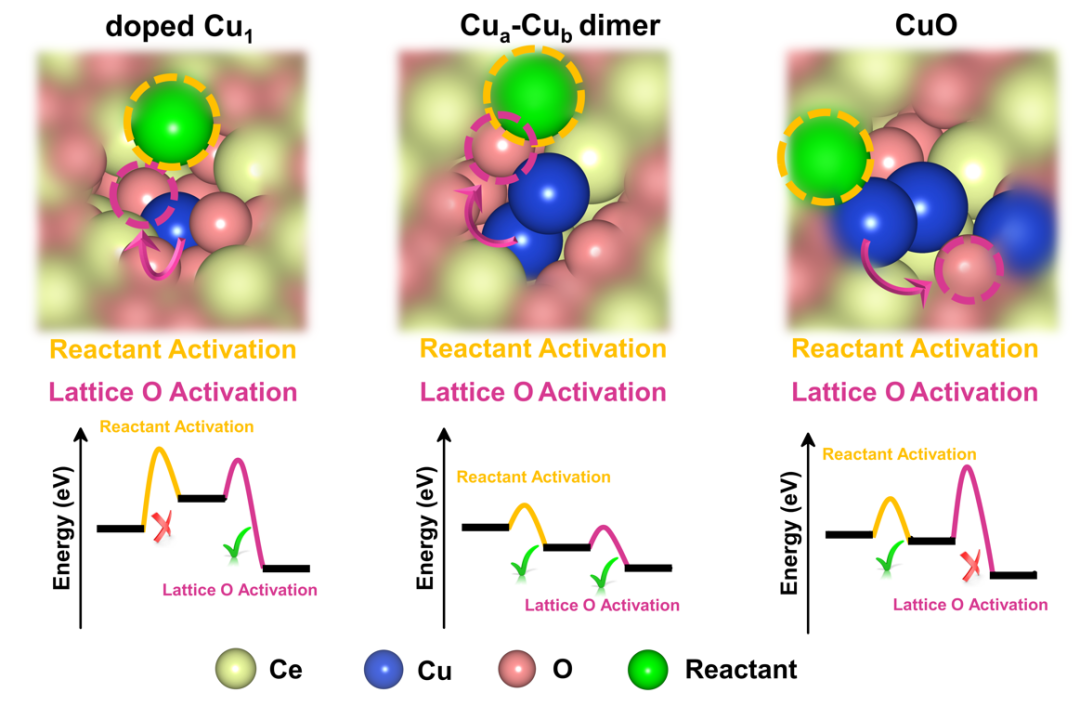
Figure 5.
Schematic illustration of the superiority of unique dimeric structure in catalytic activation in oxidation reaction.
图5. 独特二聚体结构在氧化反应中催化活化优越性说明的示意图。
Cu
a
-Cu
b
二聚体可以同时活化反应物与晶格氧,从而降低反应的动力学能垒。而单原子Cu和CuO团簇则只能单一活化晶格氧或反应物,Cu双原子中Cu
a
-Cu
b
的协同效应是提高反应活性的关键原因。










