1 制造工艺开发
1.1 原料药制造工艺开发的目的:
1.1.1 建立一个能够持续生产出符合预期质量原料药的商业制造工艺。
1.1.2 关键
Ø 持续:稳定、可控
Ø 预期质量:安全、有效
Ø 商业化:规模
1.2 传统方式
1.2.1 与原料药有关的潜在关键质量属性确立一个合适的制造工艺
1.2.2 确立一个合适的制造工艺
1.2.3 确立一个控制策略
1.3 加强方式QBD
1.3.1 评估理解和细化生产工艺
Ø 辨识物料属性和工艺参数
Ø 确定物料属性及工艺参数与CQA的关系
1.3.2 结合质量风险管理建立更优的控制策略,如设计空间的建议
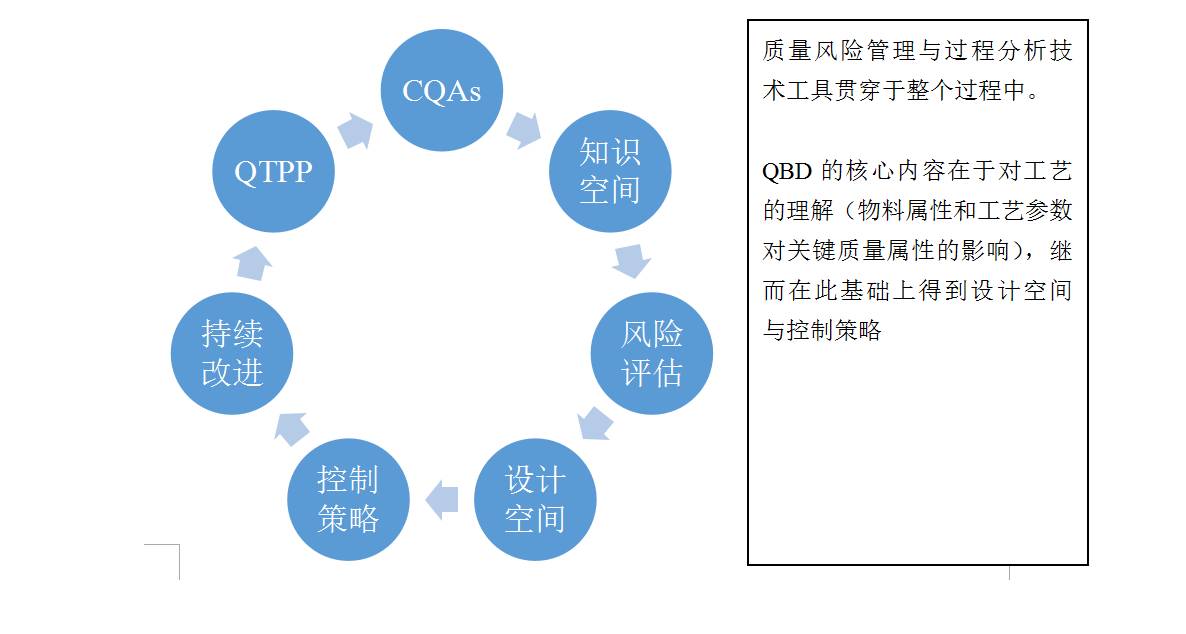
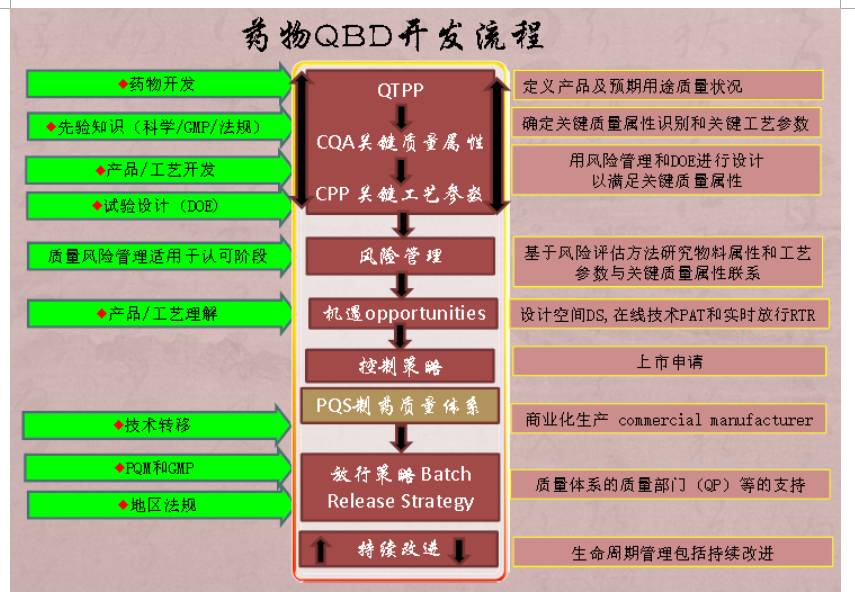
2 什么是QBD
2.1 Quality by Design (QbD): ICH Q8的定义:
A systematic approach to development that begins with predefined objectives and emphasizes product and process understanding and process control, based on sound science and quality risk management.
质量源于设计,是一种系统的研发方法,其以预先设定目标为起始,基于可靠的科学和质量风险管理,强调对产品和生产过程的理解,及对工艺的控制。
|
系统研究方法
|
以始为终,先设定目标再行动
|
|
预先设定目标
|
设定目标产品质量概况(
QTPP
)确定关键质量属性
(CQA)
|
|
产品与工艺的理解
|
确定关键物料属性(
CMA
)及关键工艺参数
(CPP)
|
|
过程控制
|
建立适宜控制策略
|
|
可靠的科学
|
利用科技文献、先进知识等
|
|
质量风险管理
|
风险评估
(ICH Q9)
|
3 什么是QBD系统
(1)确定目标产品的特性,制定产品的关键质量属性。
(2)设计满足产品CQA的物料和工艺流程。
(3)持续监控和更新工艺确保稳定的质量。
(4)确定和控制物料及工艺中变异的来源。
(5)了解物料属性和工艺参数对产品CQA的影响。
4 QBD的关键要素
|
目标产品质量概况
(
QTPP)
|
为理想地实现药物预期质量(性能)的前瞻性总结
|
|
风险评估
(Risk
Assessment)
|
在风险管理过程中组织相关信息以支持风险决策的系统化过程
|
|
关键质量属性
(CQA)
|
指某种物理、化学、生物学或微生物学的性质,应当有适当限度、范围或分布,保证预期的产品质量
|
|
关键物料属性
(CMA)
|
指某种物料(起始原料、中间体、试剂)的物理、化学、生物学或微生物学的性质,应当有适当限度、范围或分布
|
|
关键工艺参数
(CPP)
|
是指该工艺参数的变异会对产品关键质量属性产生影响,从而需要监测或控制以确保该工艺生产出预期的质量
|
|
控制策略
(Control
Strategy)
|
是指从目前的产品和过程的理解派生出的控制计划的设置,以确保工艺性能和产品质量
|
5 QBD-KEY ELEMENTS关键要素
6 目标产品质量概况
QTPP(Quality Target Product Profile)
6.1 对于API来说,QTPP一般为关注以下几点:
6.1.1 与API 化学性质相关
Ø 杂质:有机杂质\残留溶剂\残留试剂
Ø 大于定量限的杂质,不能高于原研
6.1.2 其他:符合既定药典或者ICH要求
6.1.3 含量:98.0~102.0%
6.1.4 其他指标:外观、鉴别、重金属、水分、干燥失重、炽灼残渣、等等。
7 建立目标产品质量概况的基础
7.1 原料药的合成目的:
为制剂提供质量稳定的原料药,以使其能生产出符合预期质量要求的制剂产品。
7.2 ICH (Q11):
确立原料药的预期质量时,应考虑原料药在制剂中的用途,即明确和理解对制剂产品研发产生影响的物理、化学、生物与微生物属性或特性(例如,原料药的溶解性可能影响剂型的选择)。
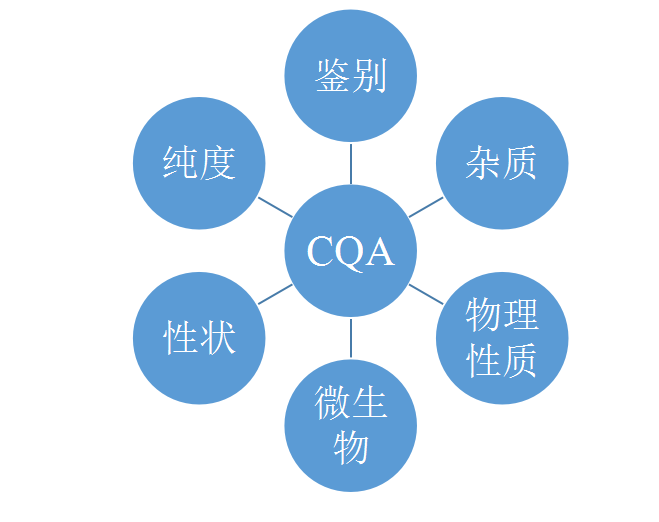
8 关键质量属性-概念
关键质量属性是一个物理、化学、生物学或微生物学属性或特征,在某个合适 的限度、范围或分布内才能确保所需的产品质量。
9
关键质量属性
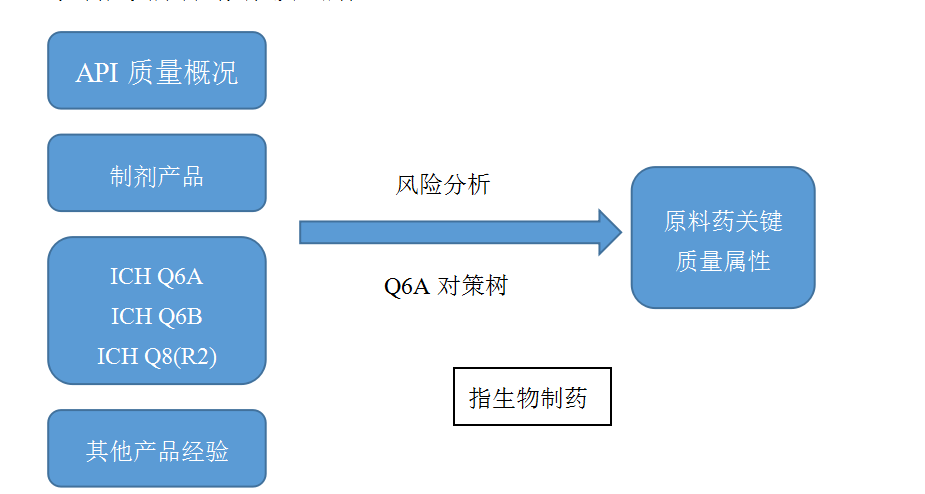
–如何定义原料药关键质量属性
9.1如何定义原料药关键质量属性:
10
与药品有关的关键质量属性
|
药品目标质量概况
|
API
关键质量属性
|
测试项目
|
关键或不关键
|
原理
|
|
鉴别
/
含量
/
均一性
|
外观描述
|
目视检测
|
Q6A
决策树
|
|
|
鉴别
|
UV/IR/HPLC
|
关键
|
正确的
API
分子结构
|
|
含量
|
含量检测
|
关键
|
药品有效性
|
|
杂质含量
|
API
杂质谱
|
有机杂质(含手性杂质)
无机杂质重金属残留溶剂
|
关键
|
药品安全性
|
|
溶出度
/
稳定性
|
晶型
/
颗粒度
|
DSC/XPRD/PSD
|
Q6A
决策树
|
药品生物等效性;稳定性
|
11
关键工艺参数
11.1 定义
工艺参数的变化影响质量属性,因此在生产过程中需要控制这些参数以保证达到预期的质量。一个关键工艺参数即使受控也仍然是关键的。因此,关键工艺参数是指那些已证实对原料药的关键质量属性有影响的参数。
11.2 工艺参数之间关系
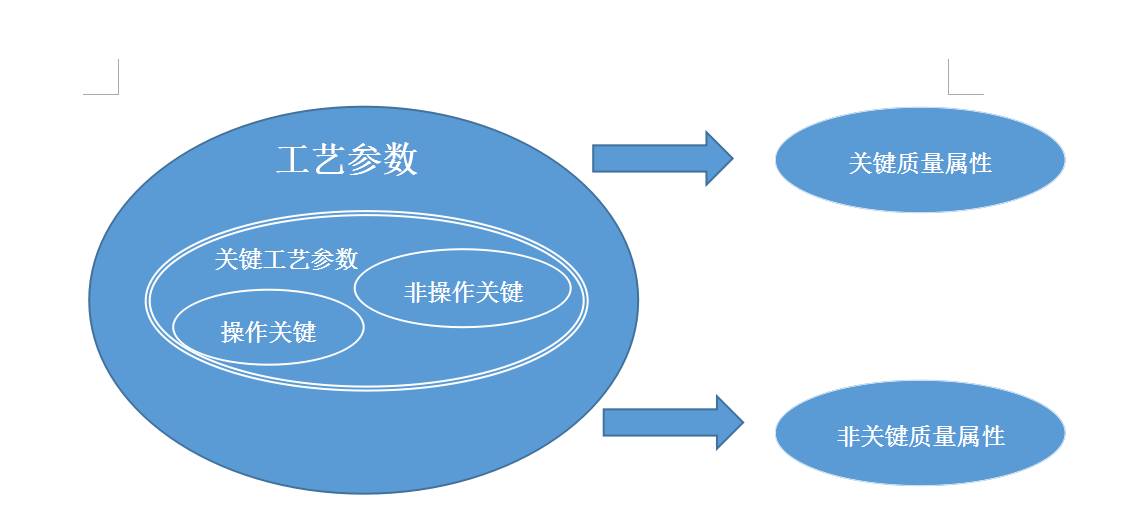
11.3 参数之间关系
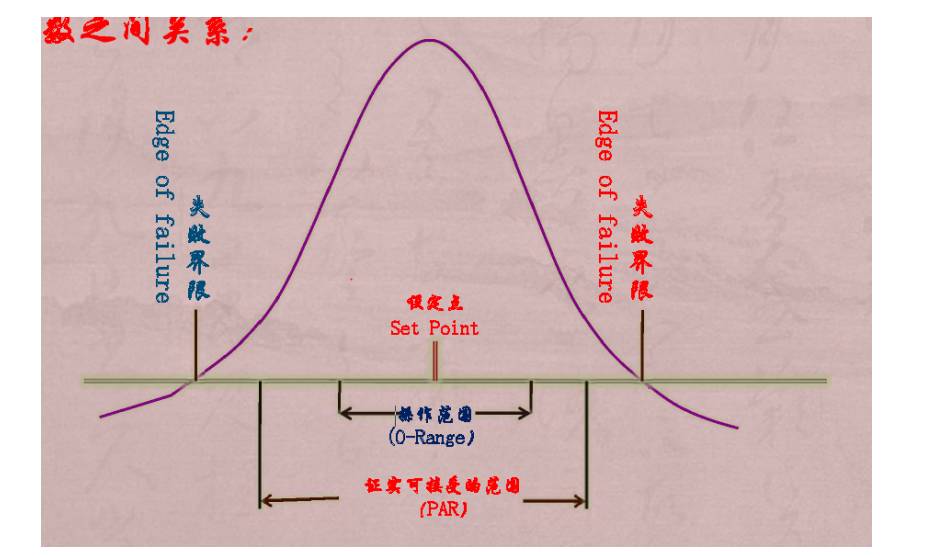
11.4操作关键参数处理
11.4.1 一旦定义为操作关键,需要采取以下措施,最好将操作标准偏差降低,以满足非操作关键的要求:
Ø 更新设备
Ø 增加自动化控制
Ø 对人员进行培训,提高操作技能
Ø 专人操作
Ø 增加双人复核
Ø 增加IPC监控
Ø 。。。
12 CMA,CPP,CQA之间的关系
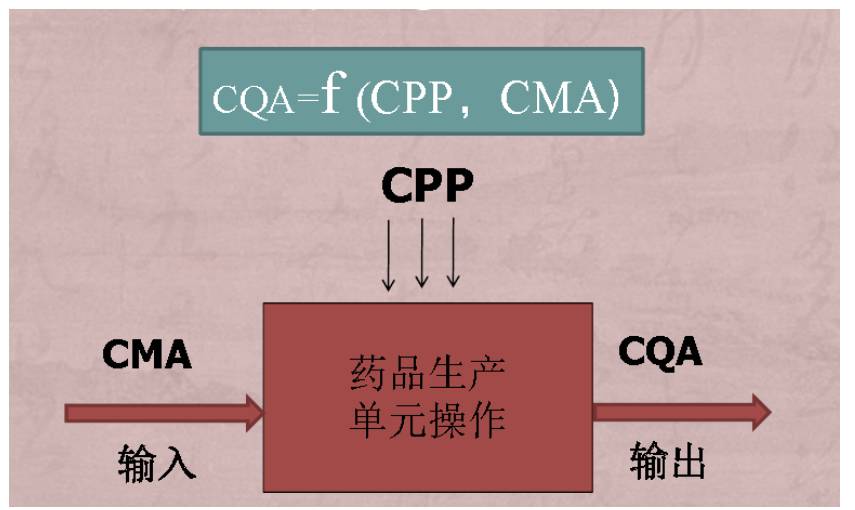
如果一种输出物料成为另一操作单元的输入物料时,它输出时的关键质量属性就可能变成输入时的关键物料属性
13
设计空间(
ICH Q8
)
²
定义:与保证质量相关的输入变量(如物料属性)和工艺参数的多种组合及其相互作用。
²
在设计空间范围内的操作不作为变更(在日常的监管中不再进行审批)。在设计空间范围之外进行的操作都算作变更,一般需由监管部门进行审批。
²
设计空间是由申请人提出,并提交给监管部门进行评估和审批。
14
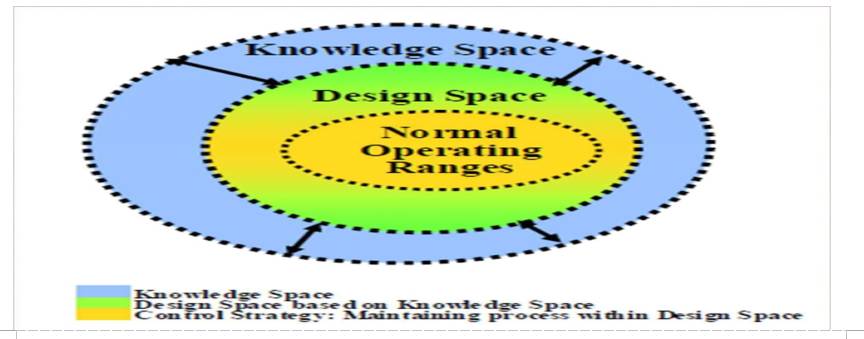
QBD
理念的关键——
设计空间
设计空间(Design space)
是已被证明有质量保障作用的物料变量和工艺参数的多维组合和交互作用
知识空间:CQAs与物料属性和工艺参数的关系
15
设计空间的确定
15.1 先申请原则
结合实验数据和化学、物理、工程等机理对产品性能进行模拟和预测
15.2 统计设计的实验
用来确定多参数的影响和它们之间相互作用的有效方法
15.3 规模化关系
一个半经验的方法,用于在不同规模(件)设备之间转化操作条件
16
设计空间
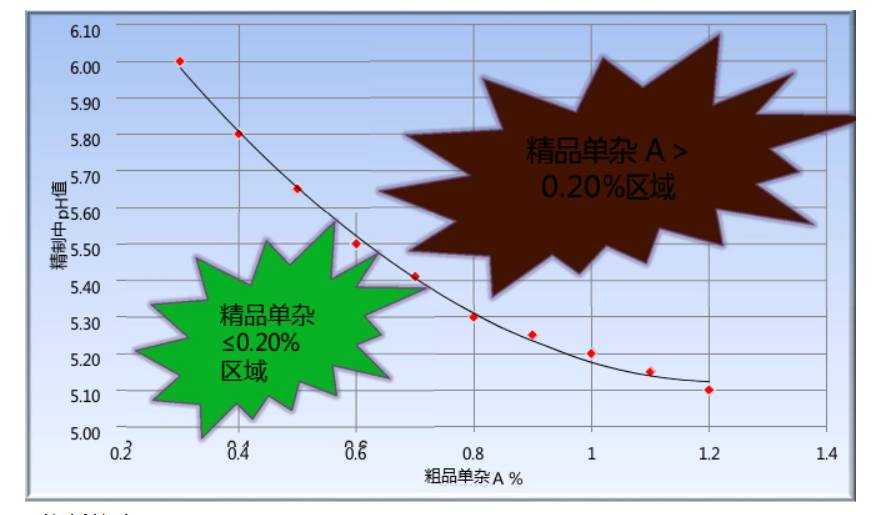
Design Space
某个最终产品单杂A的质量标准为≤
0.20%
,来源于粗品中单杂
A
,但在精制过程中可以去除一部分,主要起作用的是
pH
值,
pH
值越低,去除的能力越强,但产品收率越低,如下表所示:
|
粗品单杂
A %
|
精制中
pH
值
|
精品单杂
A %
|
|
0.3
|
6.00
|
0.20
|
|
0.4
|
5.80
|
0.20
|
|
0.5
|
5.65
|
0.20
|
|
0.6
|
5.50
|
0.20
|
|
0.7
|
5.41
|
0.20
|
|
0.8
|
5.30
|
0.20
|
|
0.9
|
5.25
|
0.20
|
|
1.0
|
5.20
|
0.20
|
|
1.1
|
5.15
|
0.20
|
|
1.2
|
5.10
|
0.20
|
u
如果采用传统方式:单纯的将两个输入变量(粗品单杂)和工艺参数固定化,如粗品单杂定义为≤
0.80%
,为了保证精品中单杂
A
≤
0.20%
,精制中pH值定义就要小于
5.30
,这样,出现两个问题:
Ø
对于粗品单杂
A
小于
0.8%
的
pH
值的过低导致收率降低很多
Ø
对于粗品单杂
A
大于
0.8%
的,从注册法规的角度讲没有办法处理,因为当初申报的粗品单杂为≤
0.8%
。
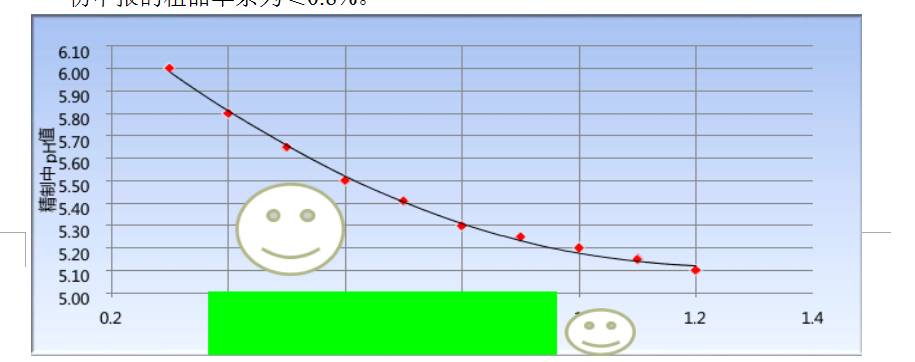
u
如果采用设计空间概念:不再将输入变量(粗品单杂)和工艺参数固定化,而是根据粗品单杂A的情况调节精制工艺中
pH
值,从而保证精品中单杂
A
≤
0.20%
。
17
控制策略
17.1 开发控制策略的考虑事项:
Ø
基于对产品和工艺的理解,保障工艺性能和产品质量的控制方式
Ø
控制策略应保证每种原料药的关键质量属性处于适当的范围、限度或分布区域。
Ø
CQA
与质量标准之间的关系如下:
ü
CQA
全部列入质量标准,通过对成品检测确认
ü
CQA
全部列入质量标准,但通过上游控制确认
ü
CQA
不列入质量标准,但通过上游控制确保
Ø
上游控制是基于对
CQA
变异来源的评估和理解
Ø
还要考虑下游因素的影响:
ü
温度变化
ü
氧化条件
ü
光照
ü
相容性等
Ø
根据风险和单一控制能力,对特定的
CQA
,在工艺中设置单一或多个控制点。
Ø
下游物料控制要严格于上游物料的控制

1.确定所有可能影响生产工艺的工艺参数和物料属性
2.利用风险评估的方法确定高风险参数和
/
或属性
3.确定这些高风险参数和属性的水平和范围
4.采用合适的
DOE
来设计实验方案
5.开展实验研究
6.分析实验数据,确定哪些工艺参数或物料属性是关键的
7.当一个工艺参数或物料属性发生实质性变化就能导致无法满足预期的关键质量属性时,该工艺参数或物料属性就是至关重要的
8.对于关键参数或属性,应限定可接受的范围(设计空间)。而对于非关键参数或属性,则可接受范围就是研究范围
 17.2 QBD理念贯穿于工艺开发全过程
17.2 QBD理念贯穿于工艺开发全过程
工艺设计阶段
• 明确目标分子的质量要求
• 设计合理路线避免杂质的生成,保证原料药质量
• 选择合适的起始原料
• 减少对环境污染,降低生产成本
工艺确认阶段
• 明确关键工艺步骤和关键工艺参数
• 进行多变量分析,进行平行正交试验
• 确定参数可控制范围
• 建立物料及产品质量标准
工艺验证阶段
• 明确规模相关参数
• 进行规模放大试验,确定参数范围
17.2.1 工艺设计阶段
(1)明确目标分子的质量概况(终产品的质量控制目标):
纯度、杂质、异构体、遗传毒性杂质、残留溶剂、晶型、粒度等。
设定依据:
Ø
已上市同(类)产品质量控制,
ICH
指导原则,前期研发经验
Ø
根据制剂(最终上市的是制剂)质量控制要求,加强原料药质量控制
|
原料药质量控制
|
与制剂质量的相关性
|
|
粒度
|
影响制剂溶出度,从而影响药物有效性
|
|
含量
|
影响制剂效力和含量均匀度
|
|
杂质:有关物质、遗传毒性杂质、残留溶剂、残留金属催化剂等
|
影响制剂杂质和稳定性
|
|
鉴别
|
制剂鉴别
|
(2)工艺路线的设计和选择:
Ø
原料和试剂的有效来源
Ø
合成步骤的最少化
与产品的质量密切相关
Ø
可能工艺相关杂质分析(原料及中间体残留、副产物、金属催化剂、残留溶剂;通过路线选择和调节尽可能减少需控制的相关杂质)
Ø
试剂及中间体的遗传毒性分析(
FDA
及欧盟相关指导原则,遗传毒性杂质需控制在
1.5ug/
天的暴露量,限度较一般杂质严格很多,通过路线选择和调节尽可能避免具有遗传毒性试剂的使用和具有遗传毒性中间体的产生)
Ø
劳动防护及环境污染
Ø
手性中心的合理引入
Ø
收率和成本的最优化
17.2.2 工艺确认阶段
确定整个生产工艺过程中的关键控制点及关键工艺参数
风险分析:
进行风险评估确定关键步骤,定义潜在的关键工艺参数,并确定参数之间的相关性。
试验设计:
进行多变量分析,确定设计空间或已证明的可接受范围。
控制策略
:
确定关键步骤、区分关键工艺参数(
CPP)
、重要工艺参数(
KPP
)和非关键工艺参数(
NCPP
);确定相关参数的控制范围。
17.2.3 工艺验证阶段
从小试规模到中试规模再到商业化规模逐级放大过程中,根据放大效应,不断调整参数控制范围,以保证产品的合格率。
试验室规模
——中试规模——试生产规模
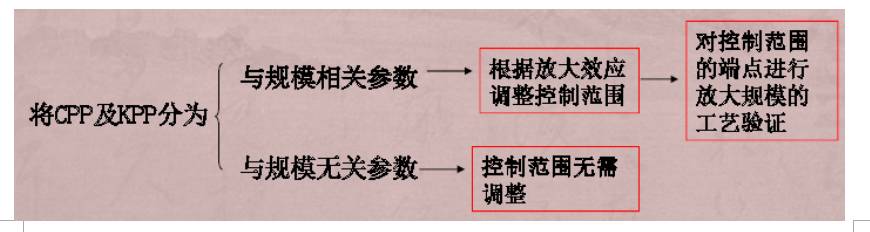
关键工艺参数(CPP)、重要工艺参数(
KPP
)
与规模相关的工艺参数

17.2.4 QBD
在工艺放大和验证中的优势
Ø
工艺参数与产品质量的相关性明确,可使关键工艺参数始终控制在可接受的范围内,提高产品质量合格的必然性,尽可能减少不合格产品的产生率。
Ø
根据参数相关性的分析,使工艺验证的目标性更加明确,减少大规模工艺验证的次数,得到理想结果。
Ø
可利用国外先进的软件和模型,对关键参数的变化进行预测,指导工艺放大研究,减少从试验室规模到商业化规模之间的逐级放大的次数,加快开发进程。
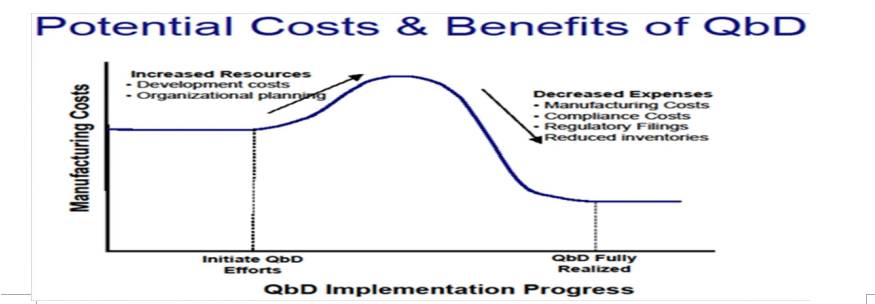
18
推行
QBD
的益处
1. 好的产品设计将减少生产过程中的问题
2. 减少上市后的变更需要递交补充申请的数量—依靠过程和风险的理解和风险缓减
3. 有助于在不需监管部门审查的情况下应用高新技术来促进生产
4. 有助于减少整体制造成本—减少浪费
5. 有助于持续性地提高或改进产品和生产工艺
6. 有助于更好地了解原料药和辅料对生产的影响
19 QB
D
的益处
本文来源:E药研发,药事纵横转载此文是觉得本文具有很高的二次分享价值,我们只用于学习与交流,如有版权问题,请联系我们删除。

药事纵横是一个开放,由自愿者组成的团体,现有成员12名,分别为Voyager88,雷诺岛,三分话,Herman,Mzwinsunny,文竹,duke,巉巉之石,ISAL,yhqqqqq,鲁礼炎,占小兵,欢迎有志之士加入我们团队。投稿、合作请加微信442015666。





