2017年6月3-4日,同写意论坛第59期活动——中国在研药品的FDA法规注册之路中,昭衍首席科学家、前美国FDA药品审评中心资深审评官姚大林博士做了关于“药物啮齿类致癌性实验研究及FDA对实验方案和结果审评的法规与科学关注点”的报告。作为一线工作人员,深有感触,也且谈谈非临床安全性评价。

姚大林博士在同写意论坛作报告
文|荣加国
药明康德副高级研究员
药物非临床安全性评价是药物研发过程中不可或缺的一个重要环节,目的在于在研新药用于人体之前以及在临床研究进行过程中,阐明靶器官的毒性反应、剂量相关性、毒性与药物暴露的关系以及毒性反应的可逆性。这些信息有助于估算首次用于人体试验的安全起始剂量和剂量范围、选择监测临床不良反应的指标,为确保临床受试者的安全提供重要的科学依据。上世纪60年代,沙利度胺不良反应事件导致上千余例婴儿出现海豹肢畸形这一惨痛教训,提醒新药进入临床实验前的安全性评价的必要性和重要性。桑国卫院士曾指出:“药物非临床安全性评价是新药研发必要阶段中极其重要的一环。”
新药研发是一个周期长、高投入、高风险的产业,一个药物要想最终能够成功上市,药物的安全性是一个非常重要的因素。毒性研究是令人敬畏的一个环节,它会对药物的安全性起到把关作用,为药物可否进入临床以及支持后续临床研究继续提供数据支持。所以,新药研发前期的安全评估极为重要,在早期介入,可以把有害化合物尽早剔除掉,为企业研发节约成本。上海新药安全评价研究中心主任马璟博士也曾说:“新药评价是个严肃的话题,要说得多么幽默、多么吸引人也不太现实”。
在进行IND递交的时候,IND package主要有以下3个方面:

非临床安全性评价主要的实验包括:一般毒性(单次/多次给药毒性试验)、生殖发育毒性试验、遗传毒性试验、安全药理试验、局部毒性试验、致癌试验、特殊毒性等(免疫原性和免疫毒性、光毒性)。非临床安全性评价在药物研发过程中需要准备的材料如下:
General template for aNME
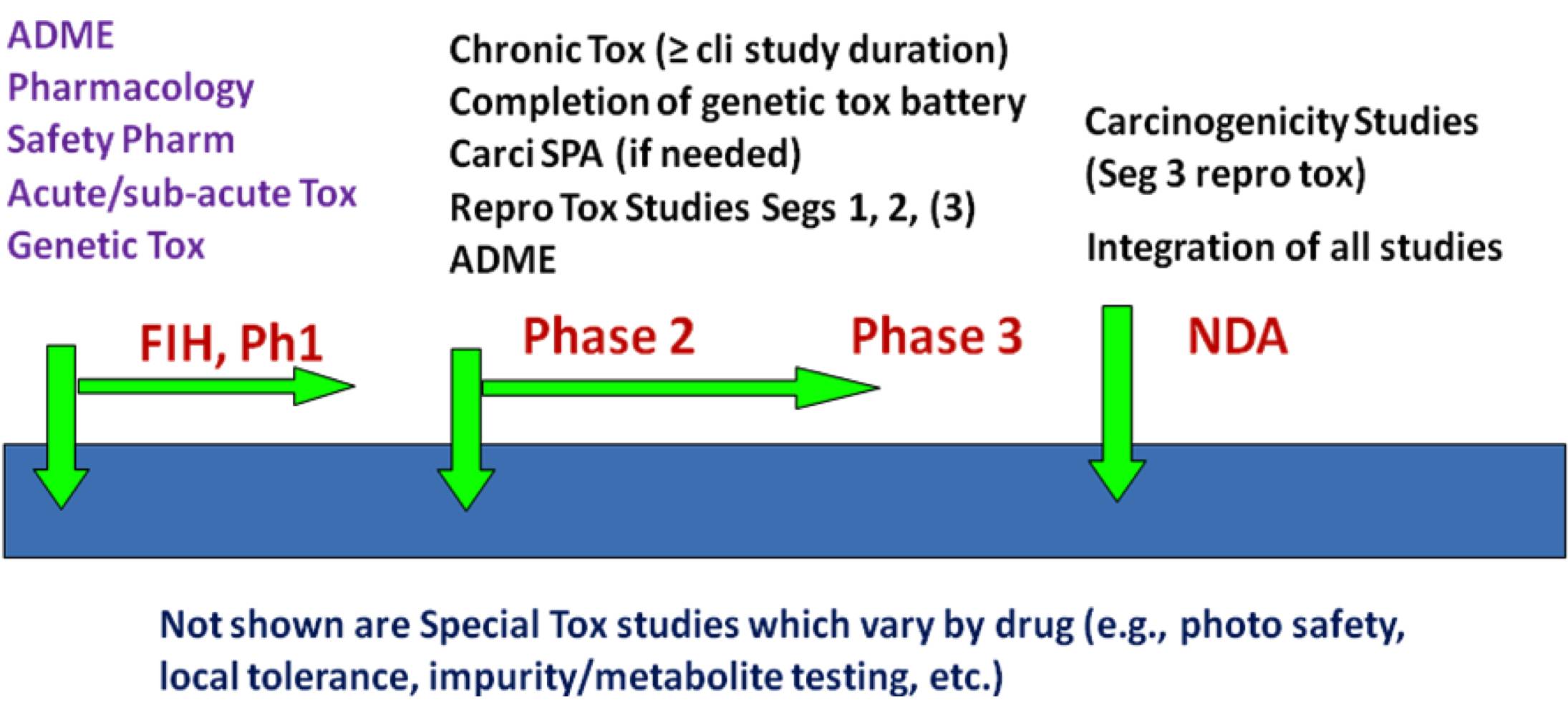
上表研究内容中除了早期研究的ADME和药理学研究不要求GLP外,所有其他项目都必须是符合GLP法规的实验研究
由于我国CFDA于今年5月31日被ICH(TheInternational Council for Harmonization)正式接纳为成员,这意味着自此药界制定技术规则的国际协调组织ICH从“三角”(美国FDA、欧盟和日本)变成了“四方”,中国药品法规机构有了话语权,与此同时,我们也必须在新药研发领域遵从ICH的技术指导原则,与国际社会全面接轨。这样一来,对药物临床前安全性评价的所有法规和技术要求也就与美国FDA的完全一致了。姚博士说:这一点上,CFDA的CDE(药审中心)行动非常迅速、高效,新出台的一系列变革举措完全因应了当前的“接轨”要求。
6月同写意的苏州专题会上,之所以把药物啮齿类致癌性实验拿出来重点讲述,是因为致癌实验算是在整个非临床安全性评价研究中最复杂、难度最大、要求最高的一个。一是耗时长,一个致癌实验一般需要连续给药2年(104周),基本涵盖了啮齿类动物的整个生命周期,加上后续的病理检查、统计学研究和发出致癌毒理报告大约需要3年时间。而一般的的毒理实验给药快则7天/14天/28天,最长不过39周。二是花费之钜,一个种属的致癌实验大约要花费125到150万美金!
需要做致癌实验的药物:1)连续用药超过6个月(如糖尿病、高血压、骨质疏松症等);2)间断性的反复用药(支气管哮喘、偏头痛等);3)延长暴露的药物(比如某些缓释药,药物在体内暴露时间较长,PK有蓄积)。此外考虑的其他因素:1)从化合物本身结构上看可能有致癌性的;2)长期慢性用药会引起组织细胞局部增生的。致癌实验一般选择以下两类:1)啮齿类动物(大鼠和小鼠)104周慢性致癌试验;2) 转基因动物(rasH2、P53等)6个月短期致癌试验外加大鼠的104周致癌试验。
开展致癌实验时间大约在整个临床研究阶段的后期,因为当做到临床III期的时候,申办方比较有把握该药可能会获批上市,更重要的是III期临床有时给药周期很长,FDA会担心有无在人体的致癌可能,尤其是临床适应症为长期用药情况下。致癌实验和III期同时做的好处是,发现问题可随时叫停或在临床上采取相应的检测手段。在实验开始前、进行中和实验结束后都要和FDA保持密切沟通。何时开展致癌试验常因药物品种而异,有些药物亦可先做一个种属,上市之后再做另一个种属的致癌实验,有的或可在上市后再开展致癌试验。因此与FDA的沟通非常重要,会起到事半功倍的效果。

实验方案设计是致癌实验的重中之重。这好比打仗时制定的作战计划,为后续的实际操作制定好了框架。实验方案设计最重要的是剂量选择,其次是给药途径和频率。剂量决定毒性、生存率、致癌性。要给大小鼠足够剂量的暴露量,观察2年期间会不会产生肿瘤以及肿瘤的类别。剂量选择方面,最常用的方法是用人体暴露量AUC的25倍作为高剂量,或者选用最大耐受剂量(MTD)作为高剂量(有时也用到其他指标,如最大可行剂量MFD或限制剂量)。中剂量和低剂量根据对数法计算。给药途径方面,建议口服给药,并选择对胃黏膜刺激最小的硅胶灌胃管进行灌胃给药。如果采用掺食给药,在药食加工过程中温度不能太高防止药物活性下降,还要考虑测PK值检测药物的暴露量。如果注射给药,应考虑到长期给药对大小鼠尾静脉血管损伤问题及可行性。整个两年给药期间,动物房的条件和对动物的护理也十分重要。
一个典型的致癌实验设计如下:1个或2个对照组+3个给药组,每组雌雄至少各60只(亦有用到65或70只每性别每组的)。在解剖的时候,一个老鼠要取60个组织,如果一个实验至少按480只老鼠算,要取多少动物组织,制备多少切片------虽然我没有做过致癌实验,但有幸见证我们机构第一个致癌实验的解剖过程,解剖人员忙碌的身影,病理部主任亲自挂帅上阵指导解剖,这场景至今历历在目。姚博士也希望FDA审评官员们要充分理解致癌实验的细节,多理解其过程的不容易,不要轻易否定企业研究结果的合理性和科学性。
致癌实验开始之前,企业要预先向FDA提交SPA (SpecialProtocol Assessment)文件。经过FDA致癌试验执行评估委员会(Executive Carcinogenicity Assessment Committee,简称eCAC)的批准才能进行致癌实验。一般在预计开展致癌实验前90天递交SPA文件,并且最好提前打招呼。SPA文件要求资料完整,并为FDA在实验开始前留有充分的时间审评和解决实验设计的有关问题。FDA审阅SPA的时限为收到文件后45天。在45天审评到期之前,eCAC内部将开会讨论,并通过投票方式决定是否同意该实验方案或给予建议。在致癌实验研究过程中,药企要和FDA保持沟通。当遇到实验后期动物大量死亡时,特别是在高剂量动物组,FDA一般建议对剩余的动物不要杀掉,剩多少算多少,并且停药,目的希望在剩余动物中观察药物在体内有没有致癌性。在这一过程中沟通、互动非常重要。致癌试验进程中的发现将对正在进行的III期临床研究产生影响,尤其在动物中发生某些特发肿瘤时。
致癌试验是药物安全性评价和上市风险控制内容的重要组成部分。国外新药的致癌性试验的建立和评价起步较早,目前对致癌性试验已经积累了大量评价研究经验,可借鉴美国FDA成熟的法规审评管理方式和国外对致癌试验的科学性的管控,对我国药品法规监管机构和新药研发企业都有重要意义。

接近一小时的演讲中,姚大林博士旁征博引,把致癌性实验讲的深入浅出,并举一些案列使得讲座妙趣横生。在会议茶歇时间正巧碰到姚大林博士,姚博士亲切的话语依然回荡在我耳边。新药研发不容易,安全评价要给力。作为一名专题负责人,深知安全评价在新药研发中的作用。还需要一步一个脚印,把安评工作做好,为新药研发尽自己的一份绵薄之力。
从了解同写意,到关注同写意,再到参加同写意活动,用了8个月的时间。正如杜新博士所言:“同写意会场布置非常有特点,把枯燥的学术活动变得诗情画意。”同写意logo映照在会场的墙壁上,像怒放着的永不凋零的花朵。在会议注册的时候,印有古诗词的笔记本,书香扑面而来;汉佛莱医药准备的会议notebook,不仅有会议目录,还有讲着介绍及内容提示;古色古香的茶杯,品茶精心,想起同写意,一切总是那么的走心,它会在你的内心深处击轻轻的击中你。在掌握知识的同时,也让我深深地记住了:同筑技术人生路,写意中国新药魂。
相关阅读
现场|一切才刚刚开始
现场|从独墅湖走到世界中心
会讯
药物研发项目的估值 6.24-25 北京






