浙大生信博士团队倾力打造的一个科研人员学习交流的公众微信平台。我们致力于科研社区服务,分享最前沿的科技进展,提供生信分析方法,解读经典分析案例,公众数据库的挖掘和临床数据统计分析。在此我们欢迎各位的加入!戳这里生信草堂公众号原文,请多关注哦~
在前面的文章中我们讨论了目前研究火热的环状RNA预测软件,通过对同一个样本采用RNAse R+/-两种建库方式,我们比较了5种常用环状RNA预测软件精确度和敏感度。在今天的推文里,我将重点选取find_circ进行详细介绍。
Find_circ工具是最早利用高通量测序数据预测环状RNA的开山鼻祖。该工具是Memczak等人2013年在权威Nature杂志上发表题为“Circular
RNAs are a large class of animal RNAs with regulatory
potency”的文章时首次发布的,从而掀起了环状RNA的研究热潮。
基于高通量测序数据的环状RNA预测的关键步骤是寻找不能连续比对到基因组或者转录组上的junction read。想要完成这项工作,第一步就是将RNA reads mapping到基因组上,然后去寻找mapping不上的序列。
Find_circ将这些mapping不上的reads各取两头20bp(保证可以唯一比对到基因组上),再次mapping到基因组上。接下来,通过短序列比对来判断GU/AG剪切位点,从而推测出潜在的环状RNA序列。图1给出环状RNA的预测过程:
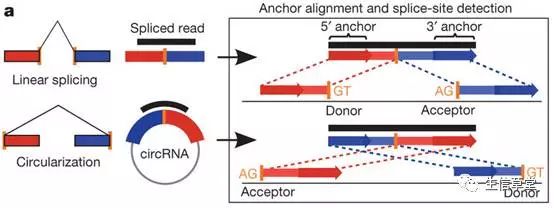
图1 环状RNA预测过程
下面给大家介绍find_circ的工作流程和命令行参数
Find_circ需要运行在装有python 2.7的64位系统上,同时需要安装numpy和pysam这两个python模块。其运行需要借助bowtie2和samtools来完成基因组mapping的过程。
基于RNA-Seq的基因组比对(pair-end模式)bowtie2
-p 16 --very-sensitive --score-min=C,-15,0 --mm -x
/path/to/bowtie2_index -q -1 mate1.fq -2 mate2.fq 2 > map.bowtie2.log
| samtools view -hbuS - | samtools sort – output.bam
###bowtie2参数介绍###
-p 使用多线程; --very-sensitive 允许多重比对,报告出最好的一个; --score-min=C,-15,0 设置比对分数函数;--mm 设置I/O模式。
###samtools view参数介绍###
-h 文件包含header line; -b 输出bam格式; -u 输出非压缩的bam格式 –S 忽略版本兼容
挑出没有比对上的序列,各取两头20bp短序列(anchor)samtools view -hf 4 output.bam | samtools view -Sb - > unmapped.bam
python unmapped2anchors.py unmapped.bam | gzip > anchors.qfa.gz
根据anchor比对基因组情况寻找潜在的circRNAbowtie2 -p 16 --reorder --mm -M20 --score-min=C,-15,0 -q -x /path/to/bowtie2_index -U anchors.qfa.gz | python find_circ.py -G /path/to/chomosomes.fa -p prefix -s find_circ.sites.log > find_circ.sites.bed 2 > find_circ.sites.reads
grep
CIRCULAR find_circ.sites.bed | grep -v chrM | gawk '$5>=2' | grep
UNAMBIGUOUS_BP | grep ANCHOR_UNIQUE | $path/maxlength.py 100000 >
finc_circ.candidates.bed
###利用grep工具筛选出符合以下条件的circRNA:
(1)线粒体染色体除外;
(2)至少有2个junction read;
(3)最大的不超过100kb
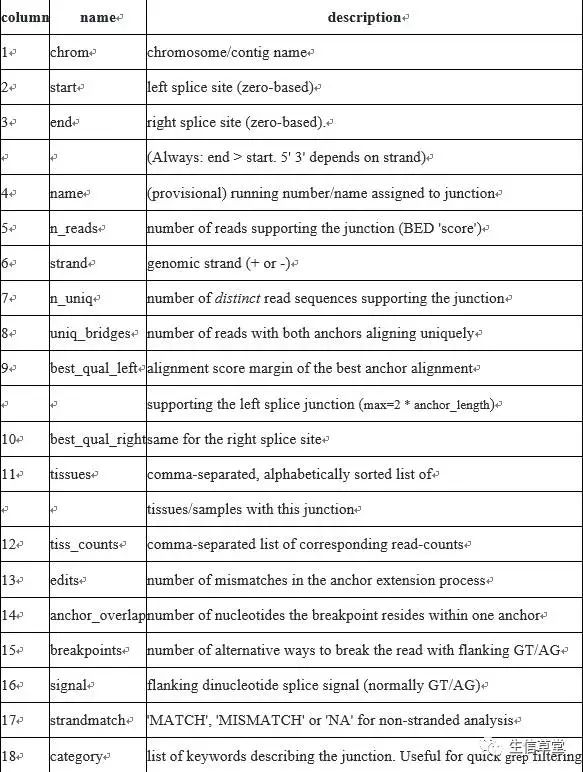
Find_circ输出BED文件格式,由18列组成,包含了预测到的环状RNA的各类指标。
关于CricRNA分析的精彩内容,我们将继续分享。
点公众号菜单里科研攻略-数据挖掘,查看往期系列~









