eCTD格式申报资料模式的探讨与展望
Discussion and Prospect of Electronic Common Technical Document Model for New Drug Application
刘晓丹1,陆 峰1,何 伍2*
(1. 上海药品审评核查中心,上海 201203;2. 国家食品药品监督管理局药品审评中心,北京 100038)
摘要:
目前国际上有超过40 个国家和地区已经采用了eCTD 标准,如美国、加拿大、欧盟及其成员国( 包括英国)、瑞
士、南非、日本等,药品注册文件递交和审评已经由纸质版过渡到了电子化。ICH M2 EWP(expert working group) 指出采用软件对电子提交的元数据和文档结构,对电子提交文件的查阅、创建文件的生命周期管理和归档等方面做了规范。为了适应新药研发的创新和国际信息化的飞跃发展,本文对CTD 格式和eCTD 格式申报资料模式的内容和流程进行了简述和概括,并结合审评工作的体会,针对大多数国家已经部分采纳或者正在逐步转向eCTD 格式申报资料模式现象,分析我国目前实际情况,尤其是新药审批若将eCTD 申报模式引入,不仅保证注册材料真实和完整,而且保证注册申报环节规范和便捷,并且对eCTD 格式申报资料模式优势进行了探讨和展望。
关键词:
CTD 通用技术文件;eCTD 格式申报;探讨与展望
药品注册申报材料一直以来都是以纸制品进行
提交,但是提交的资料越来越多,药物流通越来越频繁,这使得各个国家必须对申报资料进行统一管理。目前各国对于注册申请文件仍然没有一个统一的格式,每个国家对于提交的技术报告的组织及文件中总结和表格的制作都有自己的要求。为解决这个问题,人用药品注册技术要求国际协调会( ICH)为了协调统一ICH 地区( 美国、欧盟、日本) 注册申报资料的格式,起草了“通用技术文件”(common technical document,CTD) 进行规范。
欧盟和日本决定采用统一格式的CTD 来规范各个地区的新药注册申请,并在2003 年7 月起首先在欧洲实行。从2008 年1 月1 日开始, 美国FDA 要求采用电子CTD 格式(electronic CTD,eCTD) 进行电子申报(e-submission),所有制剂注册自2009 年6 月始必须采用eCTD 文件格式进行注册,即电子申报;新版美国处方药使用费法案则要求到2013 年实现完全的电子评审。非ICH的国家,如加拿大和澳大利亚以及世界卫生组织也均采用CTD 格式。欧洲药品管理局(EMEA) 要求从2010年1 月1 日开始采用eCTD 格式进行集中方式的申报。在国际药品注册申请的技术文件编写中,采用统一格式将会显著减少企业财力和物力的投入,缩短申请编写时间,并且简化电子递交操作。
1 CTD 的定义与组成
CTD 文件是国际公认的文件编写格式,制作
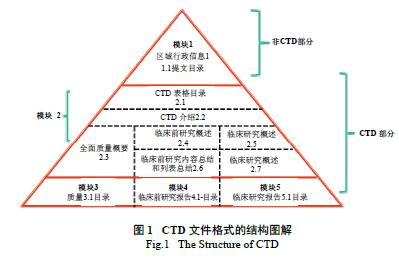
一个向药品注册机构递交的结构完善的注册申请文件,共有5 个模块,模块1 是地区特异性的,模块2、3、4 和5 在各个地区是统一的。模块1 是指地区性行政信息和法规信息,包括各地区的特殊文件,例如申请表或在各地区被建议使用的标签,其内容和格式可以由每个地区的相关注册机构来指定。模块2 是指CTD文件概述和综述,由7 个部分组成,如图1 所示,是对药物质量,非临床和临床试验方面内容的高度总结概括,必须由合格的和有经验的专家来担任文件编写工作。其中“2.1”为CTD 总目录,“2.2”为申请药品的一般介绍,主要介绍药品的药理作用及临床适应证,“2.3 ~2.7”为具有资历的专家的相关综述和概要,包括:质量概要、临床前综述、临床综述、临床前研究列表总结和表格概要、临床研究概述。模块3 是指质量部分( 药学包括原料药和制剂),提供药物在化学、制剂和生物学方面的内容。模块4 是指临床前研究报告,提供原料药和制剂在毒理学、药理学及药动学试验方面的内容。模块5 是指临床研究报告,提供制剂在各项临床试验方面的内容[1]。
2 我国出台CTD 的背景与内容
目前我国药品的供求关系已发生根本改变,药
品的可及性已非主要矛盾。医药产业“多、小、散、低”的格局没有发生根本性改变,迫切要求整合资料,提高医药行业的水平。随着国际合作的加强以及我国企业进军国际市场,均要求我们在资料上与国际统一CTD 格式。符合药品研发的规律,能更有效地展现研发过程,利于评价。而申报资料的本质是数据的提交,申报资料的结构日趋复杂化和多样化,质量参差不齐,缺乏清晰度,导致审评困难,完整审评时数据的数量和种类也需要全面掌握。
CTD 格式申报资料的核心是基于充分的研究数据和信息构建药品质量控制体系,体现过程控制( 生产工艺过程) 和终点控制( 终产品检验) 相结合的特点,提出“质量源于设计”(QbD) 的药品质量控制理念。以原料药申报资料为,“3.2.S”包括“3.2.S.1 基本信息”、“3.2.S.2 生产信息”、“3.2.S.3特性鉴定”、“3.2.S.4 原料药的质量控制”、“3.2.S.5对照品”、“3.2.S.6 包装材料和容器”和“3.2.S.7 稳定性” ;以制剂申报资料为例,“3.2.P”包括“3.2.P.1剂型及产品组成”、“3.2.P.2 产品开发”、“3.2.P.3 生产”、“3.2.P.4 原辅料的控制”、“3.2.P.5 制剂的质量控”、“3.2.P.6 对照品”和“3.2.P.7 稳定性”。我国药品审评理念也发生了巨大的变化,尤其是药学方面,更强调质量标准和重视生产工艺,引导申请企业合理全面地设计和开展研发工作,提升对产品生产过程的控制能力,而药品监管部门可对产品和





