作者:苑晓梅
责编:SXY
单细胞测序对许多复杂组织重新进行分解分析,打破了我们对细胞类型的固有认知。通常情况下,研究人员首先通过无监督聚类,获得细胞簇,然后根据Marker基因手动注释每个簇可能的细胞类型,或者应用"label transfer"比对到已经分型的数据确定自己研究的细胞类型 (这也是单细胞整合分析的一个关键点,具体关注我们的单细胞课程)。然而,对大型数据集根据Marker基因手动注释一来比较繁琐,难以扩展,二来Marker基因具有不唯一性,人为确定有选择困难症 (单细胞分群后,怎么找到Marker基因定义每一类群?)。"Label transfer"的方法需要预先注释的数据,容易受batch effects影响。
那么,就要敲黑板啦!

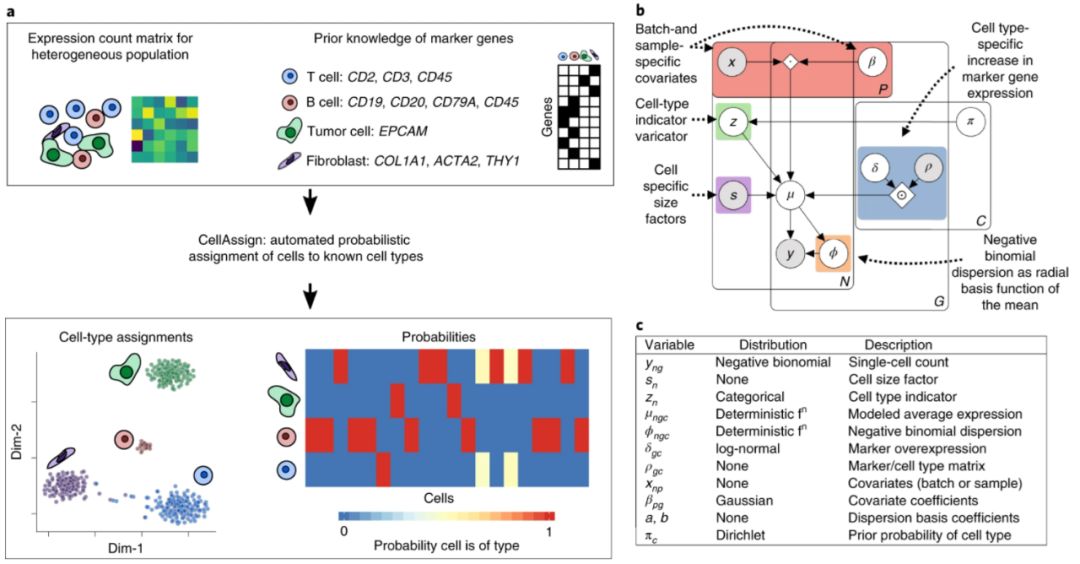
作者提出了cellassign(https://irrationone.github.io/cellassign/introduction-to-cellassign.html)-应用概率模型综合分析已知的细胞类型标记基因,将单细胞RNA测序数据注释为predefined or de novo cell types。该方法于2019年9月发表在Nature methods,原文是Probabilistic cell-type assignment of single-cell RNA-seq for tumor microenvironment profiling。
流程概览
cellassign基于Marker基因信息将单细胞RNA测序获得的细胞分型匹配到已知细胞类型。与其他从单细胞RNA-seq数据中确定细胞类型的方法不同,cellassign不需要已经标记的单细胞或bulk数据 - 只需要知道每个给定的基因是否是某种细胞类型的marker就好,想获得这些Marker基因,可以看下CellMarker数据库等。
以下为workflow (用户的输入是子图a的上半部分:标准化后的表达矩阵和Marker基因-细胞类型对照表,输出是细胞归属的已知细胞类型或新细胞类型):
包安装
cellassign的安装需要依赖于 tensorflow (机器学习经典包,莫烦Python机器学习),可以根据以下步骤进行安装:
install.packages("tensorflow")
library(tensorflow)
install_tensorflow()
安装cellassign
BiocManager::install('cellassign')
或者安装开发版
devtools::install_github("Irrationone/cellassign")
具体使用
library(SingleCellExperiment)
library(cellassign)
首先需要构建SingleCellExperiment对象,当然我们做到这步一般是已经成功构建过了,如果没有构建,可以参考以下代码:
读入数据 (Smartseq2),读入逗号或Tab键分隔的表达矩阵 (原始counts),存储为数据框,行是基因,列是细胞。
#读入逗号分隔的count matrix,存储为数据框,该数据是单细胞大名鼎鼎的小鼠全器官数据集中的一部分
dat = read.delim("FACS/Kidney-counts.csv", sep=",", header=TRUE)
dat[1:5,1:5]
## X A14.MAA000545.3_8_M.1.1 E1.MAA000545.3_8_M.1.1
## 1 0610005C13Rik 0 0
## 2 0610007C21Rik 1 0
## 3 0610007L01Rik 0 0
## 4 0610007N19Rik 0 0
## 5 0610007P08Rik 0 0
## M4.MAA000545.3_8_M.1.1 O21.MAA000545.3_8_M.1.1
## 1 0 0
## 2 0 0
## 3 0 0
## 4 0 0
## 5 0 0
数据库第一列是基因名字,把它移除作为列名字:
dim(dat)
rownames(dat) dat
构建scater对象 (更多操作见 Hemberg-lab单细胞转录组数据分析(七)-导入10X和SmartSeq2数据Tabula Muris)
sceset
# 如果做实验室记录了细胞来源的小鼠、处理等信息,可以整理成表格,采用read.table读入存储到call_anns里面一起构建scater对象`
# sceset # 查看对象
str(sceset)
作者使用一个由10个标记基因和500个细胞组成的SingleCellExperiment作为示例 (如果自己还没有数据,可以继续用作者提供的数据操作):
data(example_sce)
print(example_sce)
#> class: SingleCellExperiment
#> dim: 10 500
#> metadata(1): params
#> assays(6): BatchCellMeans BaseCellMeans ... TrueCounts counts
#> rownames(10): Gene186 Gene205 ... Gene949 Gene994
#> rowData names(6): Gene BaseGeneMean ... DEFacGroup1 DEFacGroup2
#> colnames(500): Cell1 Cell2 ... Cell499 Cell500
#> colData names(5): Cell Batch Group ExpLibSize EM_group
#> reducedDimNames(0):
#> spikeNames(0):
为了方便起见,在SingleCellExperiment的Group slot中注释了真正的细胞类型 (这里是模拟的名字,Group1,2,3等):
print(head(example_sce$Group))
#> [1] "Group1" "Group2" "Group2" "Group1" "Group1" "Group1"
关于标记基因分组,还提供了一个细胞类型与基因的二元矩阵示例(example_rho),如果基因是给定细胞类型的marker,则标记为1,否则为0:我们先从各种文献、数据库(比如CellMarker)或者直接从PanglaoDB得到先验信息,如
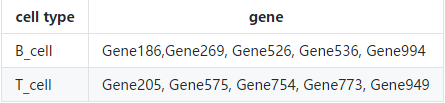
将它以列表的形式读入
example_rho T_cell = c("Gene205", "Gene575", "Gene754", "Gene773", "Gene949"))
print(str(example_rho))
#> List of 2
#> $ B_cell: chr [1:5] "Gene186" "Gene269" "Gene526" "Gene536" ...
#> $ T_cell: chr [1:5] "Gene205" "Gene575" "Gene754" "Gene773" ...
#> NULL
然后用函数marker_list_to_mat将其转换为二进制标记基因矩阵。
example_rho print(example_rho)
#> B_cell T_cell other
#> Gene186 1 0 0
#> Gene205 0 1 0
#> Gene269 1 0 0
#> Gene526 1 0 0
#> Gene536 1 0 0
#> Gene575 0 1 0
#> Gene754 0 1 0
#> Gene773 0 1 0
#> Gene949 0 1 0
#> Gene994 1 0 0
其中other表示非其中任一类型的细胞,也可用include_other=FALSE去掉该列。
表达矩阵标准化
cellassign识别的是scater对象example_sce的slots部分内容,需要用户提供量化因子用于表达矩阵的标准化。
example_sce已经预先做过运算,操作自己的数据时建议使用scran包的computeSumFactors计算,代码如下。(什么?你做的差异基因方法不合适?中提供了其它的计算方法和计算原理)
同时由于用于cell assign分析的scater对象只是原始表达矩阵的一部分,标准化时建议用原始表达矩阵所有基因进行标准化。
qclust sceset sceset
Scater对象筛选
cellassign函数需要的scater对象是单细胞实验或输入的基因表达矩阵的子集,只包含example_rho中含有的Marker gene的行;如果应用自己的数据,需要一步过滤 (example_sce测试数据已经过滤过,故跳过),代码如下(注意:Marker gene中基因的命名规则与sceset中基因命名规则一致,比如都为ENSEMBL ID或都为Gene Symbol):
# 对scater对象进行筛选
sceset
cellassign核心步骤
# cellassign object
# 获取sizefactor
s
fit marker_gene_info = example_rho,
s = s,
learning_rate = 1e-2,
shrinkage = TRUE,
verbose = FALSE)
print(fit)
#> A cellassign fit for 500 cells, 10 genes, 2 cell types with 0 covariates
#> To access cell types, call celltypes(x)
#> To access cell type probabilities, call cellprobs(x)
使用celltypes函数最大似然法估计(MLE)细胞类型:
print(head(celltypes(fit)))
#> [1] "Group1" "Group2" "Group2" "Group1" "Group1" "Group1"
以及所有MLE参数使用mleparams:
print(str(mleparams(fit)))
#> List of 9
#> $ delta : num [1:10, 1:2] 2.32 0 2.5 2.9 2.89 ...
#> ..- attr(*, "dimnames")=List of 2
#> .. ..$ : chr [1:10] "Gene186" "Gene205" "Gene269" "Gene526" ...
#> .. ..$ : chr [1:2] "Group1" "Group2"
#> $ beta : num [1:10, 1] 0.487 -0.255 -1.016 1.195 1.476 ...
#> ..- attr(*, "dimnames")=List of 2
#> .. ..$ : chr [1:10] "Gene186" "Gene205" "Gene269" "Gene526" ...
#> .. ..$ : NULL
#> $ phi : num [1:500, 1:10, 1:2] 1.86 1.87 1.86 1.86 1.86 ...
#> $ gamma : num [1:500, 1:2] 1.00 1.56e-145 1.01e-43 1.00 1.00 ...
#> ..- attr(*, "dimnames")=List of 2
#> .. ..$ : NULL
#> .. ..$ : chr [1:2] "Group1" "Group2"
#> $ mu : num [1:500, 1:10, 1:2] 22.6 80.9 11.5 15.5 15.8 ...
#> $ a : num [1:10(1d)] 1.03 1.08 1.13 1.19 1.26 ...
#> $ theta : num [1:2(1d)] 0.472 0.528
#> ..- attr(*, "dimnames")=List of 1
#> .. ..$ : chr [1:2] "Group1" "Group2"
#> $ ld_mean: num 1
#> $ ld_var : num 0.531
#> NULL
我们还可以使用cellprobs函数可视化赋值的概率,该函数返回每个细胞和细胞类型的概率矩阵:
pheatmap::pheatmap(cellprobs(fit))
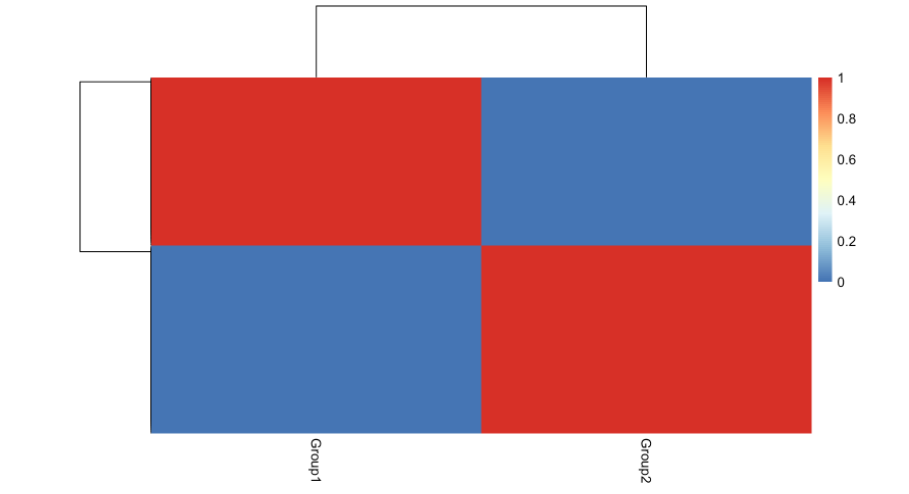
最后,由于这是模拟数据,我们可以检查与真实组值的一致性:
print(table(example_sce$Group, celltypes(fit)))
#>
#> Group1 Group2
#> Group1 236 0
#> Group2 0 264
肿瘤微环境Marker基因示例
对于人肿瘤微环境中的常见细胞类型,cellassign包中提供了一组示例标记。
标记基因可用于以下细胞类型:
这些可以通过调用进行访问:
data(example_TME_markers)
注意这是两列marker的列表:
names(example_TME_markers)
#> [1] "symbol" "ensembl"
ensembl 包含基因符号:
lapply(head(example_TME_markers$ensembl, n = 4), head, n = 4)
#> $`B cells`
#> VIM MS4A1 CD79A PTPRC
#> "ENSG00000026025" "ENSG00000156738" "ENSG00000105369" "ENSG00000081237"
#>
#> $`T cells`
#> VIM CD2 CD3D CD3E
#> "ENSG00000026025" "ENSG00000116824" "ENSG00000167286" "ENSG00000198851"
#>
#> $`Cytotoxic T cells`
#> VIM CD2 CD3D CD3E
#> "ENSG00000026025" "ENSG00000116824" "ENSG00000167286" "ENSG00000198851"
#>
#> $`Monocyte/Macrophage`
#> VIM CD14 FCGR3A CD33
#> "ENSG00000026025" "ENSG00000170458" "ENSG00000203747" "ENSG00000105383"
要将这些与cellassign一起使用,我们可以通过单元格类型矩阵将它们转换为二进制标记:
marker_mat
marker_mat[1:3, 1:3]
#> B cells T cells Cytotoxic T cells
#> ENSG00000010610 0 1 0
#> ENSG00000026025 1 1 1
#> ENSG00000039068 0 0 0
重要的是:单细胞实验或输入基因表达矩阵应该是相应的子集,以匹配标记输入矩阵的行,例如, 如果sce是具有ensembl ID作为rownames的SingleCellExperiment,则调用:# 对scater对象进行筛选
sce_marker
局限性
1.CellAssign适用于标记基因已经非常明确的条件下,未知的细胞类型或细胞状态可能是不可见的;2.作者在两种不同细胞类型中的相同标记的中等或高表达之间没有先验区分。更多单细胞操作
运行环境
sessionInfo()
#> R version 3.6.0 (2019-04-26)
#> Platform: x86_64-apple-darwin15.6.0 (64-bit)
#> Running under: macOS Mojave 10.14
#>
#> Matrix products: default
#> BLAS: /Library/Frameworks/R.framework/Versions/3.6/Resources/lib/libRblas.0.dylib
#> LAPACK: /Library/Frameworks/R.framework/Versions/3.6/Resources/lib/libRlapack.dylib
#>
#> locale:
#> [1] en_CA.UTF-8/en_CA.UTF-8/en_CA.UTF-8/C/en_CA.UTF-8/en_CA.UTF-8
#>
#> attached base packages:
#> [1] parallel stats4 stats graphics grDevices utils datasets
#> [8] methods base
#>
#> other attached packages:
#> [1] SingleCellExperiment_1.6.0 SummarizedExperiment_1.14.0
#> [3] DelayedArray_0.10.0 BiocParallel_1.18.0
#> [5] GenomicRanges_1.36.0 GenomeInfoDb_1.20.0
#> [7] cellassign_0.99.11 pheatmap_1.0.12
#> [9] matrixStats_0.54.0 edgeR_3.26.5
#> [11] org.Hs.eg.db_3.8.2 AnnotationDbi_1.46.0
#> [13] IRanges_2.18.1 S4Vectors_0.22.0
#> [15] Biobase_2.44.0 BiocGenerics_0.30.0
#> [17] limma_3.40.2 magrittr_1.5
#> [19] BiocStyle_2.12.0
#>
#> loaded via a namespace (and not attached):
#> [1] pkgload_1.0.2 bit64_0.9-7 jsonlite_1.6
#> [4] assertthat_0.2.1 BiocManager_1.30.4 blob_1.1.1
#> [7] GenomeInfoDbData_1.2.1 yaml_2.2.0 remotes_2.1.0
#> [10] sessioninfo_1.1.1 pillar_1.4.2 RSQLite_2.1.1
#> [13] backports_1.1.4 lattice_0.20-38 glue_1.3.1
#> [16] reticulate_1.12 digest_0.6.20 RColorBrewer_1.1-2
#> [19] XVector_0.24.0 colorspace_1.4-1 plyr_1.8.4
#> [22] htmltools_0.3.6 Matrix_1.2-17 pkgconfig_2.0.2
#> [25] devtools_2.0.2 bookdown_0.11 zlibbioc_1.30.0
#> [28] purrr_0.3.2 scales_1.0.0 processx_3.4.0
#> [31] whisker_0.3-2 tibble_2.1.3 usethis_1.5.1
#> [34] withr_2.1.2 cli_1.1.0 crayon_1.3.4
#> [37] memoise_1.1.0 evaluate_0.14 ps_1.3.0
#> [40] fs_1.3.1 pkgbuild_1.0.3 tools_3.6.0
#> [43] prettyunits_1.0.2 stringr_1.4.0 munsell_0.5.0
#> [46] locfit_1.5-9.1 callr_3.3.0 compiler_3.6.0
#> [49] rlang_0.4.0 grid_3.6.0 RCurl_1.95-4.12
#> [52] rstudioapi_0.10 bitops_1.0-6 base64enc_0.1-3
#> [55] rmarkdown_1.13 testthat_2.1.1 gtable_0.3.0
#> [58] DBI_1.0.0 R6_2.4.0 tfruns_1.4
#> [61] dplyr_0.8.3 knitr_1.23 tensorflow_1.13.1
#> [64] bit_1.1-14 rprojroot_1.3-2 desc_1.2.0
#> [67] stringi_1.4.3 Rcpp_1.0.1 tidyselect_0.2.5
#> [70] xfun_0.8
对单细胞转录组感兴趣的话,不妨留意一下我们将在北京召开的单细胞转录组数据整合分析专题(11月29号-12月01号):
10X登录纳斯达克,首日大涨35%,市值49亿的技术你还不会?
R统计和作图
往期精品
画图三字经 生信视频 生信系列教程
心得体会 TCGA数据库 Linux Python
高通量分析 免费在线画图 测序历史 超级增强子
生信学习视频 PPT EXCEL 文章写作 ggplot2
海哥组学 可视化套路 基因组浏览器
色彩搭配 图形排版 互作网络
自学生信 2019影响因子 GSEA 单细胞
后台回复“生信宝典福利第一波”或点击阅读原文获取教程合集












